
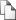
Анализ структуры хроматина и молекулярных комплексов, регулирующих транскрипцию, и распознавание функциональных элементов генома методами системной биологии
На правах рукописи
Белостоцкий Александр Александрович
АНАЛИЗ СТРУКТУРЫ ХРОМАТИНА И МОЛЕКУЛЯРНЫХ
КОМПЛЕКСОВ, РЕГУЛИРУЮЩИХ ТРАНСКРИПЦИЮ,
И РАСПОЗНАВАНИЕ ФУНКЦИОНАЛЬНЫХ ЭЛЕМЕНТОВ
ГЕНОМА МЕТОДАМИ СИСТЕМНОЙ БИОЛОГИИ
Специальность: 03.01.03 молекулярная биология
Автореферат
диссертации на соискание степени
кандидата биологических наук
Москва, 2012
Работа выполнена в лаборатории биоинформатики ФГУП «ГосНИИгенетика»
Научный руководитель:
доктор физико-математических наук,
Зав.. лабораторией биоинформатики ФГУП «ГосНИИгенетика» В.Ю. Макеев
Официальные оппоненты:
доктор биологических наук, профессор
МГУ им. М.В.Ломоносова А.А.Миронов
кандидат биологических наук
ИМБ РАН Н.Ю.Опарина
Ведущая организация: Учреждение Российской Академии Наук, Институт Проблем Передачи Информации им. А.А. Харкевича, РАН, г.Москва
Защита состоится 13 марта 2012 г. в 14 часов на заседании диссертационного совета Д 217.013.01 при ФГУП «Государственный научно-исследовательский институт генетики и селекции промышленных микроорганизмов» по адресу: 117545, Москва, 1-ый Дорожный проезд, д.1.
С диссертацией можно ознакомиться в библиотеке ФГУП «ГосНИИгенетика».
Реферат разослан « » февраля 2012 г.
Ученый секретарь
Диссертационного совета,
кандидат химических наук Т.Л. Воюшина
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ.
Актуальность темы. Анализ и распознавание регуляторных элементов ДНК, как кодирующих, так и некодирующих, представляет собой одну из основных задач вычислительной молекулярной биологии и биоинформатики. Эта область находится на стыке экспериментальной молекулярной биологии, прикладной математики и информатики. Данные о регуляторных элементах генома поставляет эксперимент, а анализ и распознавание не изученных экспериментально регуляторных участков производится с помощью специальных алгоритмов.
В настоящее время имеется крайне обширный срез методов, идентифицирующих определенные участки ДНК как регуляторные. Прежде всего это прямые методы определения участков и сайтов связывания факторов транскрипции, такие как EMSA, in vitro footprinting, in vivo footprinting, SELEX, methylation interference assay, ChIP, ChIP-chip, ChIP-seq seq (Orchard K, May GE, 1993; Galas DJ, Schmitz A; Tsai SF et al, 1991; Iguchi-Ariga SM, Schaffner W., 1989; Buck MJ, Lieb JD., 2004; Mardis ER., 2007).
Еще до появления полногеномных методов определения участков связывания белков на ДНК были сформулированы эмпирические правила того, где расположены участки связывания факторов транскрипции, какие последовательности связываются предпочтительно фактором транскрипции и как такие участки позиционированы друг относительно друга. На основании анализа результатов этих методов были построены модели мотивов, объектов, содержащих количественную информацию о наборе сайтов связывания, а именно учитывающих частоту встречаемости каждого нуклеотида в каждом положении сайта. Также выводили консенсус, то есть попросту усредненный сайт связывания для определенного фактора транскрипции или группы таких факторов. На основании такой информации производился поиск сайта связывания в геноме. При этом ставились задачи как найти уже известный сайт так и найти новые, ранее неизвестные сайты.
При проведении такого поиска оказывалось, что предсказанный сайт связывания располагался в любом месте в геноме равновероятно. Однако биологические знания говорят об обратном. Сайты связывания должны находиться в геноме крайне редко и при этом в определенных местах. С биологической точки зрения функциональные сайты связывания регуляторных белков следует искать (1) в районе, локусе, гена, (2) в определенных местах в локусе, доступных для посадки фактора транскрипции. При этом также вполне целесообразно учитывать окружение фактора, так как клеточные процессы регулируются не единичными белками, а их комплексами. Процессы в клетке, в том числе процесс транскрипции, регулируемы огромным количеством самых разных белков, обладающих разными активностями. В случае с факторами транскрипции, это разные факторы транскрипции. Они кооперативно связывают ДНК, что повышает специфичность связывания комплекса определенных участков на молекуле ДНК.
В настоящее время имеются в распоряжении методы поиска одиночных сайтов связывания фактора транскрипции, а также их плотных групп, то есть прямых повторов, палиндромов и кластеров сайтов связывания. Кластеры сайтов есть фактически комбинация прямых повторов и палиндромов с плавающим по длине спейсером. В подавляющем большинстве случаев их ищут везде в геноме, без учета специфических районов генома, в которых наличие функциональных сайтов связывания наиболее вероятно.
В этой работе установлен метод выявления предпочтительных мест связывания факторов транскрипции исходя из учета структуры хроматина. Также представлен метод, учитывающий белок-белковое взаимодействие между факторами транскрипции. Построены модели транскрипционных комплексов и выявлены динамические аспекты регуляции транскрипции.
Также приведено решение обратной задачи: распознать субъединичный состав комплекса исходя из профиля связывания субъединиц этого комплекса с ДНК.
Цели и задачи. Цель работы: анализ структуры и динамики хроматина и молекулярных комплексов и распознавание регуляторных элементов генома.
В соответствии с поставленной целью были поставлены следующие задачи.
- Разработка метода учета структуры хроматина в задачах анализа и распознавания регуляторных генома.
- Разработка метода учета белок-белкового взаимодействия в комплексе регуляторов транскрипции в задачах анализа и распознавания регуляторных участков генома и подтверждение его важнейшей роли в организации регулирующего транскрипцию комплекса.
- Анализ и распознавание структуры и особенностей функционирования транскрипционного комплекса.
- Динамическая интерпретация структур молекулярных комплексов.
Научная новизна.
1. Разработан метод учета структуры хроматина и белок-белкового взаимодействия в анализе и распознавании регуляторных элементов генома. Информация о белок-белковом взаимодействии используется как начальные данные.
2. Разработан подход учета внепиковых частей сигнала ChIP-seq, трактуемый как профиль связывания.
3. Разработана модель структуры элонгационного комплекса РНК полимеразы II с участием факторов транскрипции. Подтверждена гипотеза о стационарной транскриптосоме и предложена модель стационарной транскриптосомы.
Научно-практическая ценность работы.
Работа вносит существенный вклад в распознавание экспериментально не изученных регуляторных элементов, а также в интегральный анализ изученных. Это важно для определения, уточнения и корректировки профиля экспрессии генов и для частичной реконструкции регуляторных биологических сетей. Практические приложения безусловно могут быть полезны в таких областях, как медицина и биотехгнология. Модель биологических сетей, представленная в работе может быть полезна для понимания режима функционирования биологических сетей, что в свою очередь важно как в медицине, так и в биотехнологии.
Вклад соискателя.
1. Разработка алгоритма поиска регуляторных элементов генома исходя из данных о белок-белковом взаимодействии.
2. Анализ сигналов ChIP-seq в интерпретации профиля связывания и формулировка гипотезы о структурной связи факторов транскрипции в элонгации транскрипции с РНК полимеразой II. Обоснование гипотезы о стационарности трансркиптосомы в ядре с помощью результатов анализа сигналов ChIP-seq.
3. Введение и разработка интегральной модели биологических сетей, основанной на предположении о наличии высокой распространенности связанных колебаний в подсетях.
Апробация
Материалы диссертации были представлены на конференциях: MCCMB`09 (Москва), BGRS`10, FGD`10 (Дрезден), MCCMB`11 (Москва), SystemsX (Базель, 2011) и на совместном межлабораторном семинаре ИОГен РАН и секции молекулярной биологии ФГУП «ГосНИИгенетика».
Публикации.
По материалам диссертации опубликовано 4 статьи.
Структура и объем диссертации
Диссертация состоит из введения, аналитического обзора литературы, предложенных и разработанных методов исследования, результатов исследования и их обсуждения, выводов и списка литературы. Изложена на 150 страницах и содержит 20 рисунков и 20 таблиц.
СОДЕРЖАНИЕ РАБОТЫ
Обзор литературы изложен на 40 страницах и содержит информацию об изучении регуляции генной экспрессии, о структурах хроматина, об эволюции соответствующих методов исследования и о месте вычислительного эксперимента и моделирования в ряду этих методов.
ОБЪЕКТЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
В качестве данных, с которыми проводилась работа, были использованы координаты участков связывания факторов транскрипции, определенные крайне специфичным методом ChIP-seq и находящиеся в базе данных UCSC Genome Browser. Также были использованы данные о расщеплении ДНК ДНКазой1 и данные о положении гистоновых доменов. Эти данные были также экспортированы из UCSC Genome Browser, причем рассматривались не только процессированные результаты, но и выходные значения экспериментов в виде сигналов. Сигналы обрабатывались с помощью сервиса Galaxy, UCSC Browser Tables, Microsoft Office и программ, написанных на Perl для выявления периодичностей сигнала.
Также широко использовались программы и базы данных Genomatix и TransFac. В пределах программы TransFac наиболее интенсивно использовался инструмент Match, ищущий соответствия мотива и последовательности.
Широко применялись известные базы данных белок-белкового взаимодействия, такие как Rathway Studio, BIND, BOND, BioGrid, IntAct, String, MINT.
Обработка и анализ данных осуществлялся с помощью базы данных, созданной в среде MySQL и управляемая MySQL и специальным интерфейсем, являющимся модулем языка Perl, DBI.
Построение сетей белок-белкового взаимодействия проводилось с помощью программы String и Pathway Studio.
ОСНОВНЫЕ РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЯ
- Привлечение структурных данных для анализа и распознавания регуляторных элементов генома.
Хроматин в клетке находится в двух основных состояниях: активным и неактивном. Активный хроматин участвует в регуляции экспрессии генов, неактивный не участвует. Однако в случае распознавания регуляторных участков ДНК такой простой факт обычно не учитывается и вся ДНК рассматривается как потенциально регуляторная. Такой традиционный подход дает множество потенциально регуляторных элементов, расположенных в том числе в неактивном хроматине. Такие элементы генома могут располагаться в активном хроматине в других условиях, так как неактивный хроматин может быть преобразован в активный в клетке при определенных условиях. Распознавание при высоком пороге на качество сайта, не дает возможности адекватно определить даже небольшое количество известных регуляторных элементов, а в других случаях, при более низком пороге, определяется слишком много фрагментов ДНК как регуляторные элементы. Можно, конечно, предположить, что такая картина и является адекватной, однако адекватное распознавание сигнала включает в себя прежде всего распознавание сигнала в обучающей выборке, то есть в экспериментально изученных случаях как хотя бы не самый слабый сигнал.
Для того чтобы правильно распознавать регуляторные элементы необходимо учитывать структуру хроматина. Хроматин активный (эухроматин) отличается от хроматина неактивного (гетерохроматина) прежде всего тем, что он открытый. Такая открытость хроматина обнаруживается при расщеплении ДНК ферментом ДНКаза1. Этот фермент, являющийся по активности неспецифической эндонуклеазой, режет ДНК неспецифично относительно последовательности. Причем он режет открытые фрагменты ДНК существенно более эффективно, чем закрытые. Более того, в пределах открытого хроматина ДНКаза1 режет ДНК более интенсивно в регуляторных некодирующих областях (D Nelson, 1979). Такие области называются ДНКаза1 гиперчувствительными областями. Скажем такие элементы генома как промотор, энхансер, сайленсер являются ДНКаза1-гиперчувствительными областями.
Также для определения активности хроматина используют хроматиновые метки, такие как модификации гистонов (BD Strahl, CD Allis, 2000). Гистоны представляют собой белки с вполне компактной и плотной коровой частью и протяженными в растворитель "хвостами". Октамер гистонов состоит из плотно уложенных гистонов H2A, H2B, H3 и H4. Хвосты гистонов обращены при этом в окружающую среду. Именно эти хвосты и используются специальными гистон-модифицирующими ферментами для модификации гистонов. Наиболее модифицируемым является хвост H3 гистона. С помощью метода чип-чип (ChIP-chip), а далее и чип-сек (ChIP-seq), основанных на иммунопреципитации хроматина иммуноглобулинами против определенных белков с последующим процессированием молекулы ДНК и идентификации полученных фрагментов, были установлены участки ДНК, связанные определенным образом модифицированными гистонами.
Данные по профилю расщепления ДНКазой1 и данные по гистоновым модификациям находятся в частности в базе данных UCSC Genome Browser, где они представлены в удобном для использования виде и где имеются такие данные, полученные на разных культурах ткани, чем и обусловлен выбор этой базы данных. Эти данные использовались вместе и порознь в специально сконструированной для анализа и распознавания регуляторных областей базе данных под управлением интерфейса Perl DBI. Именно с помощью этой базы данных были установлены корреляции открытости хроматина с наличием определенных гистоновых модификаций. Далее использовались данные по участкам ДНК, определенным методом ChIP-seq как связываемые соотвествующими факторами транскрипции. Оценивалось перекрывание этих участков с гиперчувствительными к ДНКазе1 участками и участками с определенными модификациями гистонов.
Схема общего устройства базы данных приведена на Рис. 1.
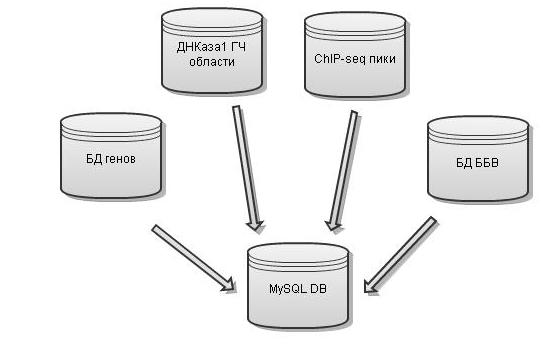
Рис. 1. Общее устройство БД определения регуляторных элементов. Информация из разных БД интегрируется в БД, в которой и осуществляется поиск участков связывания, определенных методом ChIP-seq, пересекающихся с ДНКаза1 ГЧ (гиперчувствительными) областями, между собой в случае наличия взаимодействия между соответствующими белками и с генами, регуляция которых рассматривается.
В ходе работы подтверждено наблюдение, что большинство участков связывания характеризуются пересечениями с гистоновыми доменами. Это крайне показательно, так как означает, что участки связывания факторов транскрипции находятся в часто меняющий свой статус участках. Так как распределение и профиль модификации гистонов изучаются и обнаруживаются в культуре клеток, а не в одной выделенной клетке в один и тот же момент времени для всех модификаций, то и картина получается суперпозицией профилей, наблюдаемых в данный момент времени для разных клеток, находящихся в разной фазе клеточного цикла и в разные моменты внутри одной фазы. Это можно считать еще одним подтверждением тезиса об изменчивости статуса хроматина на протяжении жизни клетки.
Также подтверждено, что подавляющее большинство участков ChIP-seq пересекаются с участками, гиперчувствительными к ДНКазе1. Понятно, что целесообразно искать сайты связывания не в геноме вообще, а в области пересечения участков связывания, определенными методом ChIP-seq, с участками, гиперчувствительными к ДНКазе1 и с гистоновыми доменами. Участки, определенные методом ChIP-seq, являются, как видно, вполне достоверными участками связывания. Поиск сайта лишь осуществляет более точное картирование связывания, если оно вообще четко локализовано.
| Название транскрипта | Хромосома | Нить ДНК | Координата 1 | Координата 2 | Координата 1 ДНКаза1 | Координата 2 ДНКаза1 | Фактор транскрипции | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15461563 | 15462956 | c-Jun | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15461563 | 15462956 | c-Jun | |
| uc010gqq.1 | chr22 | + | 15462800 | 15476000 | 15462060 | 15463192 | TAF1 | |
| uc002zlq.2 | chr22 | + | 15462800 | 15476000 | 15462060 | 15463192 | TAF1 | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15462060 | 15463192 | TAF1 | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15462060 | 15463192 | TAF1 | |
| uc010gqq.1 | chr22 | + | 15462800 | 15476000 | 15461563 | 15462956 | c-Jun | |
| uc002zlq.2 | chr22 | + | 15462800 | 15476000 | 15461563 | 15462956 | c-Jun | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15461563 | 15462956 | c-Jun | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15461563 | 15462956 | c-Jun | |
| uc010gqq.1 | chr22 | + | 15462800 | 15476000 | 15462908 | 15463188 | PU.1 | |
| uc002zlq.2 | chr22 | + | 15462800 | 15476000 | 15462908 | 15463188 | PU.1 | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15462908 | 15463188 | PU.1 | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15462908 | 15463188 | PU.1 | |
| uc010gqq.1 | chr22 | + | 15462800 | 15476000 | 15464118 | 15465431 | Ini1 | |
| uc002zlq.2 | chr22 | + | 15462800 | 15476000 | 15464118 | 15465431 | Ini1 | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15464118 | 15465431 | Ini1 | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15464118 | 15465431 | Ini1 | |
| uc010gqq.1 | chr22 | + | 15462800 | 15476000 | 15464234 | 15465490 | Brg1 | |
| uc002zlq.2 | chr22 | + | 15462800 | 15476000 | 15464234 | 15465490 | Brg1 | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15464234 | 15465490 | Brg1 | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15464234 | 15465490 | Brg1 | |
| uc010gqq.1 | chr22 | + | 15462800 | 15476000 | 15464415 | 15464983 | c-Jun | |
| uc002zlq.2 | chr22 | + | 15462800 | 15476000 | 15464415 | 15464983 | c-Jun | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15464415 | 15464983 | c-Jun | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15464415 | 15464983 | c-Jun | |
| uc002zlr.1 | chr22 | + | 15462800 | 15509720 | 15478539 | 15478685 | PU.1 | |
| uc002zls.1 | chr22 | + | 15462800 | 15559521 | 15478539 | 15478685 | PU.1 | |
Таблица 1. Пример результата работы алгоритма поиска регуляторных элементов гена. Колонки: координата транскрипта левая; координата транскрипта правая; координата ДНКаза1 сверхчувствительной области левая; координата ДНКаза1 сверхчувствительной области правая.
Из всего множества участков ChIP-seq были выбраны те, которые пересекались с участками, гиперчувствительными к обработке ДНКазе1, и те, которые пересекались с различными гистоновыми доменами. При этом рассматривались и участки, пересекающиеся как с ДНКаза1 гиперчувствительными участками, так и с гистоновыми доменами. Разработан простой алгоритм, выдающий для любого введенного названия гена или участка ДНК список экспериментально установленных регуляторных участков, пересекающихся с ним. Пример для находящегося в прицентромерной области псевдогена предположительной тирозин-форфатазы TPTE, psiTPTE22, приведен в Таблице 1.
Исследовались также участки SAR/MAR и инсуляторы на предмет пересечения с ДНКаза1 сверхчувствительными участками и гистоновыми доменами и обогащенности пересечениями с участками связывания регуляторов транскрипции, определенных методом ChIP-seq. Также разработаны приложения, позволяющие переходить от участка связывания фактора транскрипции к изучению специфической регуляции процессов и взаимодействию между различными регуляторными каскадами в пределах набора регулируемых генов.
- Учет белок-белкового взаимодействия в распознавании регуляторных элементов
Введен метод учета белок-белкового взаимодействия (ББВ) при анализе и распознавании регуляторных элементов ДНК. ББВ учитывалось на начальном этапе анализа или распознавания в соответствии со следующей схемой (Рис. 2).
Для каждого гена из базы данных UCSC Genome Browser экспортируется последовательность ДНК, представляющая локус гена. Для каждого локуса гена из базы данных UCSC Genome Browser экспортируется набор консервативных последовательностей. Эти консервативные последовательности представляют собой участки ДНК, выровненные последовательности которых были оценены программой phastCons как консервативные. Данные по консервативности используются отдельно как подключаемый модуль (подпрограмма). Локус гена исследуется на предмет наличия предполагаемых сайтов связывания факторов транскрипции. При этом используется либо стандартная программа MATCH, либо другая программа, ищущая сайты связывания. В современном варианте используется программа JASPAR. Ищутся все сайты интересующего фактора транскрипции. Для такого фактора выводится таблица взаимодействия с другими белками. Для этого используется, например, программа Pathway Studio или в современном варианте String. На основании работы с обучающей выборкой производится подбор параметров, таких как оценка качества каждого сайта и расстояние между сайтами. Эти параметры используются при анализе и распознавании сайтов. Также разработан алгоритм оптимизации поиска регуляторных элементов на основа максимизации разницы в количестве находящихся сайтов в ДНКаза1 чувствительных участках или участках ChIP-seq или в их пересечении и в контрольной выборке вне этих участков.
Самой интересной модификацией метода является алгоритм поиска белковых комплексов и регуляторных элементов на основе поиска перепредставленных в выборке указанных тестовых участков сайтов связывания факторов транскрипции, взаимодействующих с уже известным, найденным фактором транскрипции, экспериментально определенный участок которого изучается.
Архитектура представлена на Рис. 2.
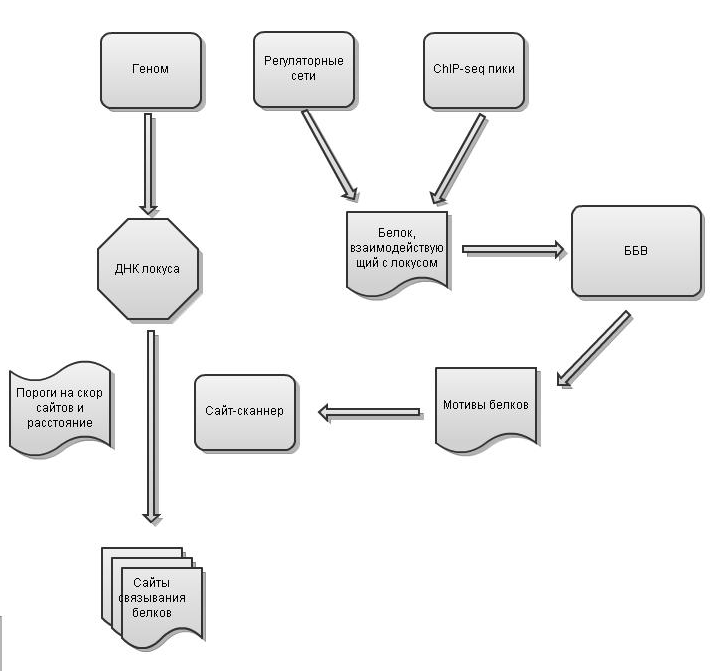
Рис. 2. Организация программы поиска регуляторных элементов на основе учета ББВ. Из генома выбирается локус ДНК, сканируемый далее набором мотивов с установленными порогами на качество соответствия мотиву каждого сайта и расстояние между ними. Набор мотивов соответствует набору белков, взаимодействующих с белков, связывающимся с локусом ДНК в случае регулируемого гена или просто по результатам метода ChIP-seq. Этап поиска групп сайтов осуществляется с помощью интерфейса Perl DBI.
Фактически в молекуле ДНК выбирались те участки, которые связывались взаимодействующими друг с другом факторами транскрипции. При этом ББВ учитывалось как источник выборки белков, сайты которых искались. Исследования для некоторых известных случаев показали, что и при этом существенно пересекались с областями, гиперчувствительными к ДНКазе1 и участками с определенными модификациями гистонов. Использовались как интегральные данные о ББВ, определенном в любых экспериментальных условиях, например в различных культурах клеток, так и ББВ с подтвержденной коэкспрессией и, стало быть, колокализующихся в определенной ткани. Видно, что учет коэкспресии имеет существенное значение. Весь алгоритм поиска групп сайтов связывания взаимодействующих белков в последовательности сайтов реализуется с помощью Perl DBI.
Рис. 3. Взаимодействие между белками, связывающими специфично ДНК. Рассматривались белки из базы данных Jaspar. Видны взаимодействующие белки, образующие кластеры с сети. Использованы методы поиска «Experiment» и «Database». Использованные вместе они дают такую картину взаимодействия. Confidence score средний 0,4.
| Программа | DiRE dcode | ||||
| E2F, Sp1 | E2F1DP1(148) | 16294 | |||
| NF1, AP2 | 16312 | ||||
| Sp1 | 16313 | ||||
| Hif1 | HIF1(180) | 16323 | |||
| Smad | 16346 | ||||
| COUP-TF1, HNF4, ER-alpha | COUP(208) | 16352 | |||
| (PPARA, RXR, RORA, ERR) | |||||
| RORA1 | 16392 | ||||
| PPARA, RXR-alpha, ER-alpha | 16396 | ||||
| YY1 | 16410 | ||||
| AP4, NF1 | NF1(295) | 16439 | |||
Таблица 2. Пример распознавания известного регуляторного элемента, являющегося даунстрим-энхансером гена EPO. Распознаны известные сайты связывания. Также распознаны и неизвестные сайты известных факторов. В первых двух колонках названия факторов транскрипции. В первой колонке распознанные заявленной здесь программой факторы, во второй факторы, распознанные широко используемой программой DiRE dcode. В третьей колонке указаны позиции относительно начала локуса EPO.
В каноническом случае для определения ББВ использовалась база данных String с отключенным Textmining и с включенными источниками данных Database и Experiment. Для поиска потенциальных сайтов связывания использовалась программа Jaspar. В наиболее продвинутом случае использовался модуль Perl. Вид сетей ББВ для всех белков позвоночных из БД Jaspar представлен на Рис. 3.
Результаты распознавания участков связывания факторов транскрипции в локусе EPO с помощью метода учета структуры хроматина с интегрированным модулем учета ББВ приведены в Таблице 2.
Из результатов не видно, что учет ББВ играет крайне важную роль в анализе и распознавании функциональных участков связывания факторов транскрипции. Фактически это означает, что специфическое ББВ либо не играет основную в организации функционального транскрипционного комплекса, что абсурдно исходя из всего опыта изучения белковых комплексов, в том числе и транскрипционных, либо базы данных содержат крайне неполную или невыверенную информацию о ББВ. Для локуса EPO с помощью программы Pathway Studio построена схема ББВ. Из нее видно насколько комплексы плотны в смысле взаимодействия. Данные представлены на Рис. 4.
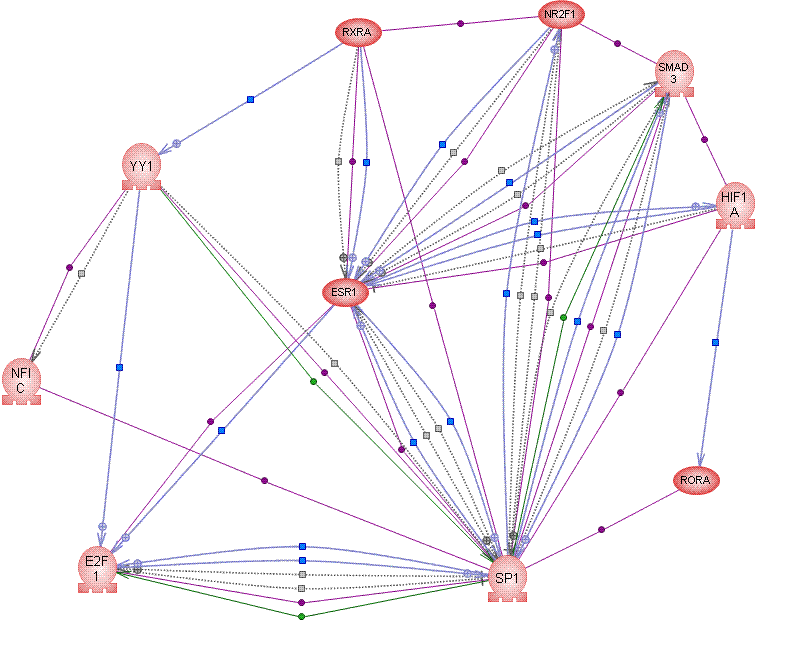
Рис 4. Модель белок-белковых взаимодействий в случае энхансера гена EPO как модельного объекта. Виден довольно плотный граф взаимодействия.
Видно, что моделируемый комплекс включает как белки, непосредственно связывающие ДНК (факторы транскрипции), как и те, которые связывают белки, связывающие ДНК (кофакторы транскрипции). Среди первых можно выделить и активаторы, и ингибиторы. Среди вторых можно выделить адапторы и гистон-модифицирующие ферменты. Такая картина встречается повсеместно.
Метод работает с существенно разной эффективностью на разных выборках. Самый оптимальный вариант это использование его на выборках участков связывания факторов транскрипции. Этот метод является скорее дескриптивным методом для определения белковых комплексов по экспериментальным участкам связывания факторов транскрипции с дальнейшим расширением регуляторных элементов.
Пример сравнения результатов по выборкам взаимодействующих и невзаимодействующих белков и попаданию в ДНКаза1 сверхчувствительные области представлен в таблице 3.
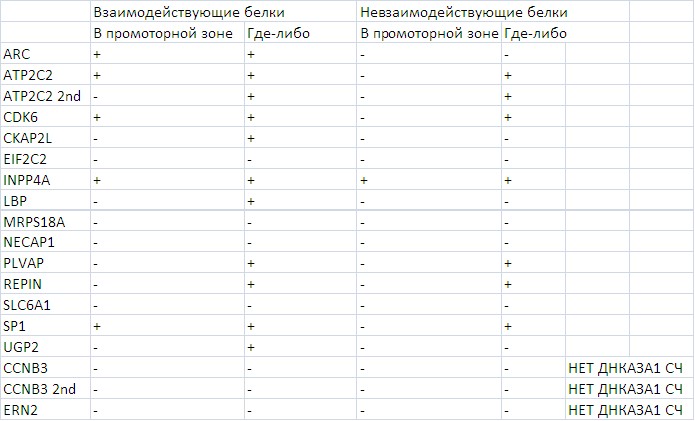 Таблица 3. Встречаемость групп сайтов взаимодействующих и невзаимодействующих белков в ДНКаза1 сверхчувствительных областях в промоторной зоне и вне ее. 2nd – условно вторая изоформа.
Таблица 3. Встречаемость групп сайтов взаимодействующих и невзаимодействующих белков в ДНКаза1 сверхчувствительных областях в промоторной зоне и вне ее. 2nd – условно вторая изоформа.
В диссертации также приведены приложения метода для анализа регуляторных элементов и SAR/MAR участков и инсуляторов.
- Гипотеза об участии факторов инициации транскрипции в элонгации транcкрипции и о стационарной транскриптосоме.
Метод ChIP-seq основан на выделении участков ДНК, коиммунопреципитирующихся с белковыми комплексами. Обычно используются пики (крупные всплески сигнала ChIP-seq, являющегося мерой связанности любого определенного участка ДНК с белком). Такие пики указывают на связывание белка в течение относительно длительного периода времени в культуре клеток именно с рассматриваемым участком ДНК. При этом понятно, что это связывание является временным в клетке, так как оно свойственно для факторов транскрипции. Однако не менее важным является анализ более кратковременного связывания, могущего быть следствием передвижения комплекса по ДНК, фактически «транзитного» связывания белка с ДНК.
Также следует учитывать то, что иммунопреципитация белкового комплекса приводит к тому, что за один белок можно выделить сшитый с ним другой белок и сшитый с другим белком участок ДНК. Этот факт играет ключевую роль в определении структуры и динамики подвижных комплексов на ДНК. При этом все эффекты диссоциации комплекса на субъединицы при проведении эксперимента, залипание на ДНК отдельных субъединиц комплексов считаются незначительными. Также считается, что специфичность антител не зависит от конформации белка и от его экранирования другими субъединицами комплекса или другими комплексами. В диссертации подробно обсуждаются такие особенности метода ChIP-seq.
Использовались сигналы связывания факторов транскрипции и РНК полимеразы II, определенные методом ChIP-seq. Они понимались исключительно как профили связывания белка на ДНК [2]. Для каждого сигнала были использованы предварительно определенные пики, определенные из сигнала процессированием. И сигнал, и пики,были загружены из базы данных UCSC Genome Browser (http://genome.ucsc.edu/). Все параметры вычислялись с помощью сервиса Tables (http://genome.ucsc.edu/cgi-bin/hgTables). Пересечения и дополнения, а также корреляции определялись с помощью коэффициента линейной корреляции в сервисе Tables.
Из-за “бага” в сервисе Tables таблица аннотированных генов UCSC knowngene экспортировалась из базы данных, нужные колонки txStart и txEnd вместе с колонкой chrom вырезались из таблицы и импортировались обратно в базу данных как custom track. Экзоны генов из выборки knowngene обрабатывались таким же способом. В таком виде поиск пересечений осуществлялся адекватно. Пересечения сигнала с выборкой аннотированных генов и означают участки сигнала, попадающие в гены, условно говоря “сигнал в генах”.
В действительности, широко использовались все данные, и пиковые, и внепиковые, и их совокупность. Однако, для доказательной части работы пиковые данные не необходимы и поэтому не представлены, некоторые из них лишь упомянуты.
Были выбраны все регуляторы транскрипции, для которых имелись данные ChIP-seq, определенные на культуре HeLa в пределах проекта Encode (http://genome.ucsc.edu/ENCODE/), а именно AP-2a, AP-2g, c-Fos, c-Jun, c-Myc, E2F1, E2F4, E2F6, HA-E2F1, junD, Max, Nrf1, TR4, BAF155, BAF170, Ini1, Brg1, BDP1, BRF1, BRF2, RPC155, TFIIIC. Функции этих белков различны. Факторы транскрипции с AP-2a по TR4 включительно, выбранные для метода ChIP-seq субъединицы знаменитого Swi/Snf комплекса ремоделирования хроматина с BAF155 по Brg1 и выбранные факторы инициации транскрипции РНК полимеразы III, который использовался как контроль, с BDP1 по RPC155. Также использовались самые разные данные из других культур клеток.
Рассматривалась самая маленькая аутосома человека, 22-ую хромосома. В случае рассмотрения очень больших массивов данных ставился фильтр на количество значений 10 MB. Также рассматривались урезанные выборки по 100 KB для выяснения тенденции в эффекте при увеличении выборки. Сборка генома hg18.
3.1 Регуляторы транскрипции, осуществляемой РНК полимеразой II, связываются с внутригенными областями, и особенно с экзонами, эффективнее, чем с внегенными областями
Уровень внепикового сигнала ChIP-seq оказывается существенно больше внутри генов и особенно внутри экзонов, нежели вне их. Результаты показаны на Рис. 5. Учитывая более медленное транскрибирование экзонов, это крайне интересное наблюдение, свидетельствующее в пользу участия факторов транскрипции в элонгации. Однако это еще не есть критерий специфического участия факторов транскрипции в элонгации и структурной связи с РНК полимеразой II. Однако в пользу этого говорит корреляция сигналов ChIP-seq.
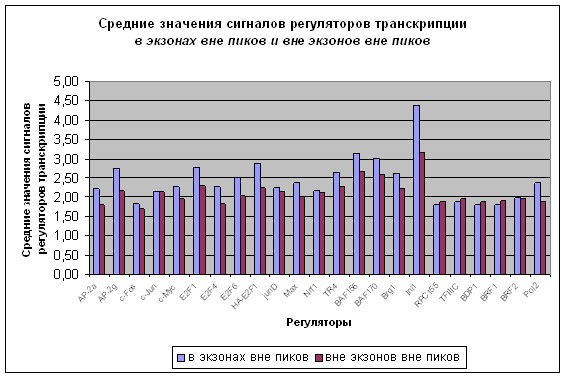
Рис. 5 Средние значения сигналов регуляторов транскрипции в экзонах вне пиков и вне экзонов вне пиков для указанных регуляторов.
3.2 РНК полимераза II и факторы транскрипции в генах, и, в частности в экзонах, связывают предпочтительно одни и те же участки ДНК
Сигналы регуляторов транскрипции проявляют существенную положительную коррелированность с сигналом РНК полимеразы II. Регуляторы РНК полимеразы III, транскрибирующей гены некодирующих РНК, адекватно выступают в качестве контроля. Результаты показаны на Рис. 6.
Корреляция сигналов факторов транскрипции РНК полимеразы II и сигнала самой РНК полимеразы II примерно такая же, как и корреляция сигналов белков Swi/Snf комплекса и сигнала РНК полимеразы II. Комплекс Swi/Snf считается не только ассоциированным с РНК полимеразой II на промоторе (что наблюдается и по геномным данным, данные не представлены), но и мигрирующим вместе с РНК полимеразой II в процессе элонгации. Таким образом, есть основания считать факторы транскрипции как минимум мигрирующими с РНК полимеразой II в процессе элонгации транскрипции (M. A. Schwabish, K. Struhl, 2007), что уже подразумевает по крайней мере опосредованную структурную связь между ними и РНК полимеразой II в процессе элонгации транскрипции. 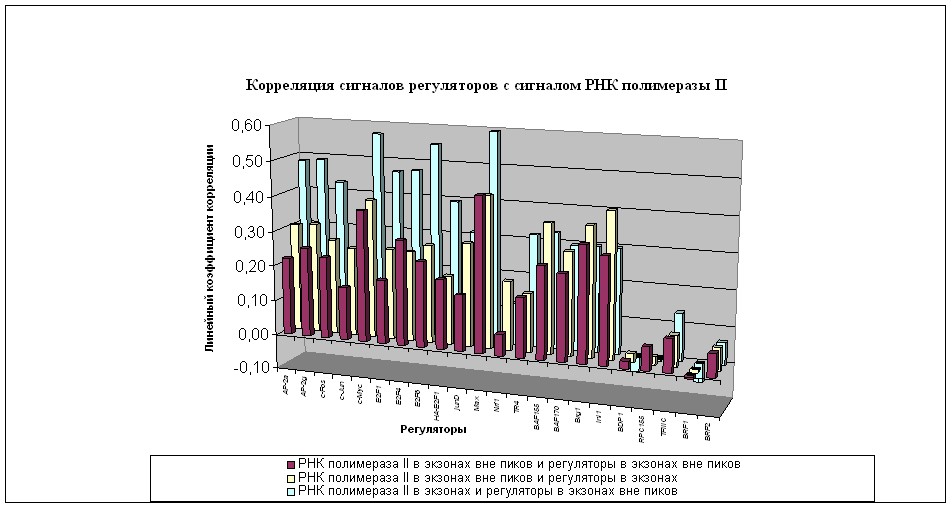
Рис. 6 Корреляция сигналов регуляторов с сигналом РНК полимеразы II. Линейный коэффициент корреляции указанных в легенде типов сигналов ChIP-seq в экзонах.
Корреляция между сигналом регулятора транскрипции и сигналом РНК полимеразы II ткань-специфична. Корреляция между сигналами регулятора РНК полимеразы II из культуры HeLa и сигналом РНК полимеразы II из культуры GM12878 существенно менее выраженная, чем корреляции в каждой из указанных клеточных линий.
РНК полимераза II не только колокализована в существенной степени с факторами транскрипции и хроматин-ремоделирующим комплексом, но и в существенной степени колокализуется с ними на промоторах генов. Для целей проверки пересечения пиков РНК полимеразы II с промоторами была сконструирована база данных с таблицей известных генов и таблицами пиков РНК полимеразы II в разных культурах клеток. Искались пересечения пиков РНК полимеразы II с концами генов с учетом ориентации гена по цепи ДНК. Оказалось, что 64% пиков РНК полимеразы из культуры клеток HeLa пересекаются с промоторами известных генов. Из этих пиков 87% пересекаются с BAF155. Таким образом, РНК полимераза II предпочтительно колокализуется с комплексом ремоделирования когда находится на промоторе.
При этом установлено, что площадь под сигналом РНК полимеразы II на промоторе больше, чем площадь этого же сигнала на протяжении всего гена. Сделан вывод о том, что этот эффект связан либо с тем, что РНК полимераза II находится в инициации дольше, чем во всей элонгации, либо РНК полимераза II всегда находится в области промотора, будучи связанной через комплекс с ДНК даже в процессе элонгации.
- Связь структуры комплексов и динамики процессов в них.
Выдвигается гипотеза, что почти все процессы в клетке проявляют осцилляторный режим функционирования. Предполагается, что те процессы, которые протекают иначе, в свою очередь вынуждены подстраиваться под динамику основных, колебательных процессов. Такой вывод можно сделать исходя из самой структуры биологических сетей и из моделирования этих сетей. Биологические сети на молекулярном уровне представимы, формально выражаясь, системами с огромным количеством обратных связей, из которых имеется существенная часть негативных. В таких сетях происходят либо единичные всплески активности, либо периодические изменения активности. Причем чем сильнее негативная обратная связь, тем больше вероятность возникновения осцилляций в активности белков, в экспрессии регулируемых этими белками генов и соотвественно в активности продуктов генов. Минимальный замкнутый контур с негативной обратной связью представляет собой «цикл». В работе было проведено выделение и разграничение отдельных циклов в некоторых модельных системах, указана связь между этими циклами. Была предложена модель, в которой циклы с высокочастотными колебаниями соответствуют непосредственно взаимодействующим в комплексе белкам. Циклы с низкочастотными колебаниями предполагаются соответствующими системам со структурно разделенными компонентами. Учитывая, что все белки в клетке так или иначе находятся в составе каких-либо комплексов, предполагается, что относительно низкочастотные колебания свойственны процессам передачи информации в клетке, будь то экспрессия генетической информации на всех уровнях или будь то каскады клеточной сигнализации. Предполагается, что все высокочастотные циклы могут проявлять связанные колебания и даже участвовать в поддержании низкочастотных колебаний, порождающих, например циркадные ритмы или, возможно, клеточный цикл.в низкочастотные на основании пространственного расположения в клетке.
В подтверждение гипотезы были проанализированы сети ББВ, показывающие взаимодействия между белками, считающимися непосредственными регуляторами циркадных ритмов. Показана некоторая обособленность этой подсети в сети ББВ в клетке. Сделано предположение, что такая изоляция необходима для поддержания условно автономного режима функционирования этой подсети. При этом фактор транскрипции NFkB, активность которого осциллирует в клетке (Nelson DE et al., 2004), взаимодействует с многими другими белками из самых разных систем клетки. Причастность других белков к разным регуляторным сетям и каскадам была установлена. Таким образом, можно считать это пусть не доказательством, но некоторым свидетельством в пользу вовлечения участников самых разных регуляторных подсетей в одну большую сеть с осцилляторным режимом функционирования.
Пример сети ББВ с участием RelA/p65 приведен ниже, на Рис. 7.

Рис. 7. Сеть ББВ с участием осциллирующего RelA/p65.
Исходя из этого, построена циклическая модель сетей и выведена соответствующая гипотеза относительно цикличной структуры сетей и группового осцилляторного характера их активности. Разработана модель представления метаболических, сигналинговых, генных сетей в виде системы концентрический окружностей, каждая из которых представляет собой цикл. Самая общая модель такой сети представлена на Рис. 8.
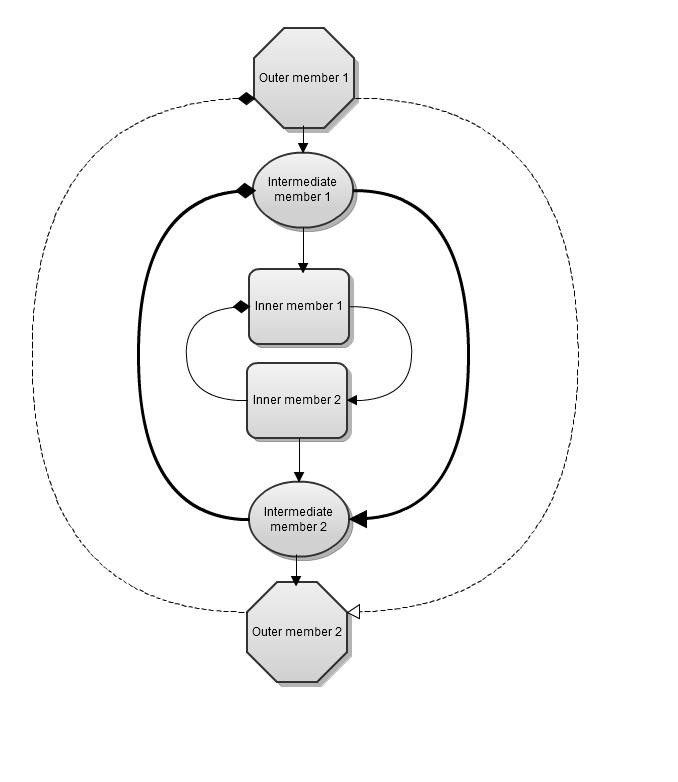
Рис. 8. Самая общая модель циклических сетей. Стрелками обозначены прямые позитивные связи (активация), ромбами обозначены обратные негативные связи (ингибирование). Внутренний цикл относится к высокочастотным колебаниям, промежуточный – к среднечастотным, а внешний – к низкочастотным. Есть все основания предположить, что внутренний цикл это метаболические сети, промежуточный это генные сети и сигналинг, а внешний это циркадные ритмы или даже клеточный цикл.
Рассмотрены экспериментальные факты, подтверждающие, что для такого функционирования сети более чем достаточно аргументов. Разобраны примеры колебаний в метаболических, в генных и в сигналинговых сетях.
Колебания в сигналинговых сетях коррелируют по периоду с колебаниями в регулируемых ими генных сетях. Период осцилляций сигналинговых сетей и генных сетей кратен периоду осцилляции в метаболизме. Если учесть эти факты, то понятно, что можно представить все биологические сети на молекулярном уровне как систему циклов, связанных в ключевых узлах, и обеспечивающих согласованные периодические изменения активности ферментов и регуляторов их активности синтеза.
Если учесть, что некоторые белки, отвечающие за самые общие функции в клетке, связаны функционально с белками, регулирующими циркадные ритмы, то можно сделать вывод о том, что такая слаженная система циклов представляет собой «шестеренки» для клеточных «часов».
ЗАКЛЮЧЕНИЕ
Разработан комплексный метод поиска регуляторных участков генома на основании анализа геномных данных по связыванию белков, данных о структуре хроматина, данных по модификациям гистонов в локусе и на основе данных о ББВ. Привлечение данных о ББВ позволяет подходить вплотную как к моделированию белковых комплексов, так и к моделированию регуляторных элементов генома. Использование этого алгоритма представляет интерес для фундаментальной биологии, а также для биотехнологии, медицины.
Разработан подход к учету внепикового сигнала ChIP-seq как профиля связывания белков на ДНК и к учету структуры комплексов при анализе сигнала ChIP-seq. Исходя из анализа профиля связывания белков в гене было предположено участие факторов транскрипции в элонгации и построена модель транскриптосомы и предположена ее стационарность в ядре. При этом использовался внепиковый сигнал ChIP-seq и интерпретация его как сигнала «транзитного» связывания.
Введенная гипотеза о цикличности биологических сетей молекулярного уровня позволила дать интерпретацию важности устойчивого функционирования ферментов, в том числе РНК полимеразы II, в составе объемных комплексов, состоящих из десятков субъединиц.
ВЫВОДЫ
- Сконструирован, программно реализован и верифицирован алгоритм, учитывающий структуру хроматина в задачах анализа и распознавания регуляторных элементов генома.
- Разработан, программно реализован и верифицирован алгоритм, учитывающий белок-белковое взаимодействие в задачах анализа и распознавания регуляторных элементов генома.
- Предположена сложная структура молекулярного комплекса, регулирующего элонгацию транскрипции и включающая в себя как комплекс ремоделирования хроматина и гистон-модифицирующие ферменты, так и факторы транскрипции, развивается гипотеза о стационарных транскрипционных комплексах.
- Дана динамическая интерпретация важности сборки большеразмерных комплексов в клетке исходя из разработанной гипотезы о модулярной цикличности биологических сетей молекулярного уровня и о колебательной динамике таких сетей.
СПИСОК ПУБЛИКЦИЙ
1. Alexander Belostotsky, Conception of biological networks at the molecular level as orchestrated systems of oscillators representing interconnected modular molecular clocks, Journal of Metabolomics and Systems Biology, Vol. 2(2), pp. 15-19, September 2011
2. Белостоцкий А.А., Анализ профиля связывания белков с ДНК, определенного методом ChIP-seq, выявляет возможное взаимодействие специфичных факторов транскрипции с РНК полимеразой II в процессе элонгации транскрипции, Биофизика, Т.57, N 2, с.359-365
3. Kulakovskiy IV, Belostotsky AA, Kasianov AS, Esipova NG, Medvedeva YA, Eliseeva IA, Makeev VJ. A deeper look into transcription regulatory code by preferred pair distance templates for transcription factor binding sites. Bioinformatics. 2011 Oct 1;27(19):2621-4. Epub 2011 Aug 18.
4. Кулаковский И. В., Касьянов А. С., Белостоцкий А. А., Елисеева И. А., Макеев В. Ю., Предпочтительные расстояния между участками ДНК, связывающими белковые факторы, регулирующие инициацию транскрипции, Биофизика. - 2011. - Т. 56, N 1. - С.136-139.