
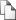
Молекулярно генетическое изучение разнообразия и микроэволюции yersinia pestis
На правах рукописи
Платонов
Михаил Евгеньевич
Молекулярногенетическое изучение
разнообразия и микроэволюции Yersinia pestis
03.02.03 – микробиология
03.01.03 – молекулярная биология
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата биологических наук
Оболенск - 2010
Работа выполнена в Федеральном государственном учреждении науки «Государственный научный центр прикладной микробиологии и биотехнологии» Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека
Научные руководители:
доктор медицинских наук, профессор Анисимов Андрей Павлович;
кандидат медицинских наук Дентовская Светлана Владимировна
Официальные оппоненты:
доктор биологических наук, профессор Шемякин Игорь Георгиевич;
доктор биологических наук, профессор Попов Юрий Алексеевич
Ведущая организация: Федеральное государственное учреждение здравоохранения «Иркутский научно-исследовательский противочумный институт Сибири и Дальнего Востока» Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека
Защита состоится «20» октября 2010 г. в 12 часов на заседании диссертационного совета Д 350.002.01 при Федеральном государственном учреждении науки «Государственный научный центр прикладной микробиологии и биотехнологии» Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека Российской Федерации по адресу: 142279, Московская обл., Серпуховской р-н, п. Оболенск.
С диссертацией можно ознакомиться в библиотеке Федерального государственного учреждения науки «Государственный научный центр прикладной микробиологии и биотехнологии»
Автореферат разослан «20» сентября 2010 г.
Учёный секретарь диссертационного совета
кандидат биологических наук Н.К. Фурсова
Общая характеристика работы
Актуальность проблемы. Чума – острое инфекционное заболевание, относящееся к группе особо опасных карантинных инфекций. Возбудитель чумы, Yersinia pestis, - самый опасный из бактериальных патогенов. Чума была причиной трех пандемий и привела к гибели более 200 миллионов человек. Эта природно-очаговая инфекция передается укусами кровососущих насекомых – блох и циркулирует в популяциях диких грызунов, но при легочной форме может передаваться непосредственно от человека к человеку [Perry, Fetherston, 1997]. Помимо чумного микроба, род Yersinia включает в себя еще 14 видов [Souza et al., 2010], два из которых - Y. pseudotuberculosis и Y. enterocolitica, вызывают у человека заболевания желудочно-кишечного тракта [Brubaker, 1991]. Данные молекулярно-биологических исследований указывают на то, что вид Y. pestis произошел от Y. pseudotuberculosis O:1b серотипа [Skurnik et al., 2000] в последние 1500-20000 лет [Achtman et al., 2004]. Природные очаги чумы имеются на всех материках, кроме Австралии и Антарктиды, занимая 6-7 % территории земного шара [Perry, Fetherston, 1997]. На территории бывшего Советского Союза насчитывается 42 природных очага чумы, 11 из них - в Российской Федерации [Онищенко, Кутырев, 2004]. Следствием географической и физической разобщенности природных очагов, влияния различных биотических и абиотических факторов, явилась адаптация, в результате которой чумной микроб приспособился к циркуляции в популяциях более 200 видов диких грызунов и лагоморфов, используя около 120 видов блох в качестве переносчиков [Онищенко, Кутырев, 2004]. В процессе аллопатрического видообразования сформировались подвиды, различающиеся по своим питательным потребностям, способности ферментировать различные субстраты и вирулентности для чувствительных к ним млекопитающих. До недавнего времени вид Y. pestis подразделяли на шесть подвидов: pestis, altaica, caucasica, hissarica, talassica и ulegeica [Anisimov et al., 2004]. По биохимической активности внутривидовые группы Y. pestis делили на три биовара: antiqua, mediaevalis, orientalis [Devignat, 1951].
Для типирования Y. pestis применяют как фенотипические, так и генетические методы. В основу фенотипических методов положены питательные потребности, способность ферментировать различные субстраты, и вирулентность для различных видов животных. Хотя фенотипические методы и позволяют различать подвиды и биовары Y. pestis, с их помощью невозможно проводить дифференциацию на уровне штаммов. К тому же нестабильность проявления фенотипических признаков, их зависимость от условий эксперимента, а также наличие атипичных штаммов, существенно затрудняют интерпретацию результатов [Апарин, Голубинский, 1989; Anisimov et al., 2004].
В связи с этим, решающую роль в типировании чумного микроба, играют молекулярно-биологические методы. В разное время с этой целью использовали плазмидный скрининг [Балахонов, 1989; Filippov et al., 1990], определение полиморфизма длин рестрикционных фрагментов – RFLP (Restriction Fragments Length Polymorphism) [Picard, 2000], риботипирование [Guiyoule et al., 1994], IS-типирование [Бобров, 1995; Motin et al., 2002; Torrea et al., 2006], и PCR-фингерпринт. К методам PCR-фингерпринта относятся случайно амплифицированные полиморфные ДНК RAPD-PCR (Randomly Amplified Polymorphic DNA) [Huang et al., 2000], повторяющиеся экстрагенны палиндромы – REP-PCR (Repetitive Extragenic Palindromic sequences) [Kim et al., 2003] и повторяющиеся межгенные консенсусные последовательности энтеробактерий ERIC-PCR (Enterobacterial Repetitive Intergenic Consensus sequences) [Kingston J.J., et al., 2009]. Общими недостатками этих методов являются: низкая межлабораторная воспроизводимость, необходимость в большом количестве материала для исследования, сложность использования полученных результатов для создания совместимых баз данных.
Доступность полной нуклеотидной последовательности геномов микроорганизмов [Parkhill et al., 2001] послужила толчком для развития новых современных молекулярно-генетических методов. В настоящее время для генотипирования Y. pestis широко используют многолокусный анализ вариабельных тандемных повторов – MLVA (Multiple Locus Variable-Number Tandem Repeats Analysis) [Klevytska et al., 2001; Le Flche et al., 2001], DFR-типирование – анализ отличающихся участков ДНК (Different Region), CRISPR-типирование (Clustered Regularly Interspaced Short Palindromic Repeats – кластеризованные короткие палиндромные повторы, разделенные спейсерами) [Pourcel et al., 2005]. SNP-типирование – анализ полиморфизма единичных нуклеотидов (Single-Nucleotide Polymorphism) [Schork et al., 2000], полногеномный сиквенс [Eppinger et al., 2010]. Данные методы лишены перечисленных выше недостатков и обладают высокой разрешающей способностью.
Активизация в конце XX века ряда природных очагов чумы Африки, Азии и Америки послужила причиной увеличения заболеваемости людей бубонной и легочной формами данной инфекции, что указывает на нестабильность эпидемиологической обстановки [Онищенко и др., 1998]. Не меняется эта тенденция и в начале XXI века [Онищенко, Кутырев, 2004]. Возрастание числа эпидемических проявлений чумы, наряду с возможностью перемещения авиатранспортом инфицированных Y. pestis людей в течение нескольких часов с одного континента на другой, а также возрастающая опасность международного терроризма свидетельствуют об актуальности совершенствования молекулярно-эпидемиологических методов, направленных на генетическую паспортизацию штаммов Y. pestis из различных природных очагов, необходимую для выявления источников заражения и проведения адекватных лечебных и санитарно-эпидемиологических мероприятий. В условиях сложившейся эпидемической ситуации развитие и использование новых методов в клинической диагностике и эпидемиологических исследованиях приобретает особое значение. В качестве наиболее перспективных, следует выделить молекулярно-биологические методы, особенно комплексное сочетание нескольких методов [Shangkuan et al., 1997; Sander et al., 1998], что позволит более точно выявлять звенья эпидемиологических цепочек распространения инфекции и своевременно локализовать очаги чумы.
Изучение разнообразия и микроэволюции Y. pestis важно как для решения фундаментальных задач медицинской микробиологии, таких как выявление механизмов происхождения и эволюции инфекционных болезней человека и животных, так и для разработки новых высокоспецифичных методов молекулярной эпидемиологии, а именно идентификации, гено- и геномотипирования Y. pestis. Генотипирование – один из наиболее эффективных и относительно недорогих методов, применяемых для целей паспортизации природных очагов, эпидемиологического расследования случаев заболеваний и экспертизы актов биотерроризма.
Цель исследования - изучение генетического разнообразия и микроэволюции чумного микроба из природных очагов Российской Федерации и сопредельных государств методами DFR- и MLVA-типирования.
Задачи исследования:
1. Создать пополняемый электронный каталог "геномных портретов" (DFR- и MLVA25-профилей) представительного набора штаммов Y. pestis, включающий данные, полученные в нашей лаборатории, лабораториях коллабораторов: Gilles Vergnaud (Universit Paris-Sud, Institut de Gntique et Microbiologie, Orsay, France) и Ruifu Yang (Laboratory of Analytical Microbiology, State Key Laboratory of Pathogen and Biosecurity, Institute of Microbiology and Epidemiology, Beijing, China), а также информацию о штаммах с доступными в Интернете полными последовательностями геномов.
2. Провести DFR-типирование штаммов Y. pestis; оценить дискриминирующую способность метода, корректность кластеризации внутривидовых групп и возможность использования для оценки микроэволюции.
3. Провести многолокусный VNTR анализ штаммов Y. pestis; оценить дискриминирующую способность метода, корректность кластеризации внутривидовых групп и возможность использования для оценки микроэволюции; сравнить эффективность MLVA25- и DFR-типирования.
4. Подготовить предложения по дополнению внутривидовой таксономии Y. pestis и привидению ее в соответствие с правилами Международного кодекса номенклатуры бактерий.
Научная новизна. Обнаружено 60 новых DFR-генотипов и 352 новых MLVA25-генотипов штаммов чумного микроба. Установлено, что на территории одного природного очага чумы может циркулировать несколько DFR-, MLVA25- и DFR/MLVA25-генотипов, входящих в единый генетический кластер. В то же время одинаковые DFR-типы могут входить в состав разных MLVA25- и DFR/MLVA25-кластеров, что свидетельствует о возможности гомоплазии по DFR-профилям. Циркуляция филогенетически близких MLVA25- и DFR/MLVA25-генотипов на территориях смежных природных очагов свидетельствует о целесообразности их объединения. Так, Присеванский горный (5) и Зангезуро-Карабахский горный (6) очаги должны быть объединены в Нагорно-Закавказский (5/6), Бозчельский равнинно-предгорный (8) и Джейранчельский равнинно-предгорный (11) – в Бозчельско-Джейранчельский равнинно-предгорный (8/11), а к Горно-алтайскому очагу (36) – присоединены соответствующие территории Монголии. Кластеризация на основе DFR- и MLVA-профилей соответствует таковой при SNP-типировании, а сочетанный анализ результатов DFR- и MLVA-методов повышает их разрешающую способность и позволяет получать филограммы, свидетельствующие о следующем порядке дивергенции в ходе микроэволюции возбудителя чумы: Y. pseudotuberculosis Y. pestis bv. antiqua (африканская ветвь) Y. pestis subsp. angola Y. pestis subsp. caucasica Y. pestis bv. intermedium Y. pestis bv. antiqua (азиатская ветвь) Y. pestis bv. medievalis Y. pestis bv. orientalis Y. pestis subsp. ulegeica Y. pestis subspp. qinghaiensis, talassica, xilingolensis, hissarica и altaica.
Практическая значимость и внедрение результатов работы. В GeneBank депонировано 8 последовательностей VNTR-локусов (международный уровень внедрения).
В коллекции микроорганизмов ГНЦ ПМБ депонирован набор из 10 штаммов Y. pestis - типовых для многолокусного VNTR анализа (федеральный уровень внедрения).
Создан постоянно пополняемый электронный каталог "геномных портретов" (DFR- и MLVA25, включающий на момент написания диссертации информацию о 601 штамме Y. pestis.
Установлено, что выделенные на территории Монголии штаммы подвида altaica входят в состав двух разных ветвей кластера talassica/qinghaiensis/xilingolensis/hissarica/altaica, один из которых представлен штаммами близкими штаммам подвида altaica, циркулирующим на территории Горного Алтая в России. Второй кластер представлен штаммами подобными штаммам подвида xilingolensis, циркулирующим в L очаге на территории Китая. Необходима проверка штаммов подвида altaica в коллекциях учреждений, проводящих работы с возбудителем чумы для определения их действительного таксономического положения.
Изучение штаммов Y. pestis методами DFR- и MLVA-типирования рекомендовано нами в качестве одного из дополнительных критериев внутривидовой классификации возбудителя чумы.
Результаты проведенных исследований послужили основой или были учтены при составлении методических рекомендаций "Генотипирование штаммов Yersinia pestis методом мультилокусного VNTR-анализа". Оболенск, 2009 (учрежденческий уровень внедрения). Материалы диссертации используются в лекциях для магистрантов Пущинского государственного университета и аспирантов ГНЦ ПМБ, а также при обучении современным методам генотипирования специалистов противочумных учреждений.
Положения, выносимые на защиту:
1. Разработанный комплекс методических приемов (электронный каталог, включающий информацию о DFR- и MLVA25-генотипах 601 штамма Y. pestis, а также методические рекомендации по проведению молекулярного типирования методом MLVA25) позволяет определять принадлежность штаммов чумного микроба к конкретным подвидам и биоварам, а также к природным очагам чумы: 1, 4, 5-7, 8/11, 9, 10, 31/33, 34, 36, 37, 38, 39, 40/M, 43, расположенным на территории стран СНГ.
2. Сочетание универсальной вирулентности со способностью ферментировать рамнозу, а также DFR/MLVA25-профиль, занимающий промежуточное положение между таковыми у представителей азиатской ветви bv. antiqua и кавказской группой штаммов bv. microtus, позволяет выделить штаммы, циркулирующие в очаге B Китая в новый внутривидовой таксон - биовар intermedium.
3. Сочетанное использование DFR/MLVA25-типирования позволяет предложить следующий порядок дивергенции в ходе микроэволюции возбудителя чумы: Y. pseudotuberculosis Y. pestis bv. antiqua (африканская ветвь) Y. pestis subsp. angola Y. pestis subsp. caucasica Y. pestis bv. intermedium Y. pestis bv. antiqua (азиатская ветвь) Y. pestis bv. medievalis Y. pestis bv. orientalis Y. pestis subsp. ulegeica Y. pestis subspp. qinghaiensis, talassica, xilingolensis, hissarica и altaica.
4. Несоответствие правилам Международного кодекса номенклатуры бактерий внутривидовой таксономии чумного микроба требует переименования биовара microtus в подвид microtus, а входящих в него в настоящее время филогенетических групп, а именно подвидов altaica, angola, caucasica, hissarica, qinghaiensis, talassica, ulegeica и xilingolensis – в биовары altaica, angola, caucasica, hissarica, qinghaiensis, talassica, ulegeica и xilingolensis.
Работа выполнена в лаборатории микробиологии чумы отдела особо опасных инфекций (ЛМЧ) ГНЦ ПМБ по планам НИР в рамках проекта МНТЦ № 2426 (научный руководитель - к.м.н. С.В. Дентовская); проекта РФФИ № 08-04-00405-а (научный руководитель - д.м.н., проф. А.П. Анисимов) и Госконтрактов № 119-Д от 11.06.2009 г. (№ гос. регистрации 01890017743) и № 53-Д от 29.06.2010 г. с Роспотребнадзором (научный руководитель - д.м.н., проф. А.П. Анисимов).
Личный вклад соискателя. Экспериментальные результаты, представленные в диссертации, получены лично автором в сотрудничестве, главным образом, с В.В. Евсеевой (ЛМЧ ГНЦ ПМБ). На защиту вынесены только те положения и результаты экспериментов, в получении которых роль автора была определяющей. В исследованиях также принимали участие сотрудники ЛМЧ ГНЦ ПМБ Е.В. Чиркова и Т.В. Гапельченкова. Всем им автор выражает глубокую благодарность. Кроме того, следует отметить вклад в выполнение данной работы к.м.н. С.В. Дентовской (ЛМЧ ГНЦ ПМБ) и д.м.н., проф. А.П. Анисимова (ГНЦ ПМБ) за интересное и полезное обсуждение результатов на всех ее этапах.
Апробация работы. Результаты диссертационной работы были представлены, доложены и обсуждены на: NIAID Research Conference (Opatija, Croatia, 2006); VII-й Межгосударственной научно-практической конференции государств-участников СНГ (Оболенск, 2006 г.); 9th International Symposium on Yersinia (Lexington, Kentucky, USA, 2006); II Всероссийской научно-практической конференции с международным участием "Инфекции, обусловленные иерсиниями" (Санкт-Петербург, 2006); научно-практической конференции "Современные аспекты эпидемиологического надзора за особо опасными инфекционными заболеваниями на Юге России" (Ставрополь, 2007); 9th Symposium of Analytical Microbiology (Beijing, People’s Republic of China, 2007); NATO Science for Peace and Security Programme Advanced Research Workshop “Emerging and Endemic Pathogens: Advances in Surveillance, Detection, and Identification” (Тбилиси, Грузия, 2008); научно-практической конференции молодых ученых и специалистов научно-исследовательских учреждений Роспотребнадзора "Биологическая безопасность в современном мире" (Оболенск, 2009); научно-практической конференции "Современные аспекты эпидемиологического надзора и профилактики особо опасных и природно-очаговых болезней", посвященной 75-летию Иркутского ордена Трудового Красного Знамени научно-исследовательского противочумного института Сибири и Дальнего Востока (Иркутск, 2009); научно-практической школе-конференции молодых ученых и специалистов научно-исследовательских организаций Роспотребнадзора "Современные технологии обеспечения биологической безопасности" (Оболенск, 2010); 10th International Symposium on Yersinia (Recife, Brazil, 2010); International Symposium on Bacterial Evolution and Pathogenesis (Zhenjiang, Jiangsu province, China, 2010).
Публикации. Основное содержание работы отражено в 9 научных публикациях (три статьи в международных рецензируемых журналах, реферируемых ISI).
Объем и структура диссертации. Диссертация изложена на страницах машинописного текста и состоит из введения, обзора литературы, результатов и обсуждения, заключения, выводов и списка литературы, включающего 39 работ отечественных и 102 работы зарубежных авторов. Работа иллюстрирована 16 рисунками и 10 таблицами.
СОДЕРЖАНИЕ РАБОТЫ
Материалы и методы исследования
В работе использовано 176 штаммов Y. pestis, которые выращивали на агаре Хоттингера с добавлением 1 % гемолизированной крови в течение 48 ч при температуре 28 °С. Геномную ДНК выделяли в соответствии с методическими указаниями МУ 1.3.1794 – 03 «Организация работы при исследованиях методом ПЦР материала, инфицированного микроорганизмами I-II групп патогенности» с использованием комплекта реагентов для выделения ДНК из биопроб производства НПФ «Литех».
В филогенетическом анализе также использовали данные по DFR- и MLVA25-типам 403 штамма чумного микроба, предоставленные зарубежными коллабораторами: Gilles Vergnaud (Франция) и Ruifu Yang (Китай). Опубликованные полногеномные последовательности 22 штаммов чумного микроба, а так же штамма Y. pseudotuberculosis IP32953 использовали для генотипирования методами мультилокусного VNTR анализа и DFR-анализа in silico.
DFR-анализ штаммов Y. pestis проводили, как описано ранее [Zhou et al., 2004b; Dai et al., 2005]. Полученные результаты вносили в базу данных программы Bionumerics 5.1. Для построения дендрограммы использовали метод Neighbor-Joining с коэффициентом Dice для бинарных данных. При построении дерева в качестве корневого вида использовали штамм Y. pseudotuberculosis IP32953. Для того чтобы определить DFR-генотипы профили исследованных штаммов сравнивали с профилями 32 геномоваров, выявленных ранее [Li et al., 2008].
MLVA25 типирование штаммов Y. pestis проводили, как описано ранее [Le Flche et al., 2001] в модификации [Li et al., 2009]. Полученные результаты вносили в базу данных программы Bionumerics 5.1. Кластерный анализ выполняли с использованием метода Neighbor-Joining и категорического коэффициента. При построении дерева в качестве корневого вида использовали штамм Y. pseudotuberculosis IP32953.
Анализ и сравнение результатов полученных при использовании различных методов генотипирования проводили с помощью опции "composite data set" программы Bionumerics 5.1. О дискриминирующей способности методов DFR- и MLVA25-типирования по отдельности, а также сочетанного анализа DFR/MLVA25 судили по индексу разнообразия (Din) E.H. Simpson’а [Simpson, 1949], рассчитанному, как было описано ранее [Hunter, Gaston, 1988].
Созданный и постоянно пополняемый электронный каталог штаммов Y. pestis и их генотипов представляет собой электронную таблицу в формате Microsoft Excel. Вся информация по одному штамму представлена в ячейках одной строки (возможно внесение до 256 параметров): номер штамма, видовое название, время выделения, место выделения/природный очаг, источник выделения/основной хозяин, подвид, биовар, а затем следуют ячейки с генотипами. При введении в таблицу данных генотипирования в первую ячейку вносят номер генотипа, а в последующие ячейки, число которых соответствует количеству анализируемых локусов, нумерические данные результатов исследований: для DFR-типирования – "1" или "0" при наличии или отсутствии амплификата, соответственно, а для MLVA – количество тандемных повторов в VNTR-локусе или "0" при отсутсвии амплификата. Таблица Microsoft Excel может быть импортирована в программу Bionumerics 5.1 в соответствии с рекомендациями разработчиков (www.applied-maths.com).
Результаты исследования
На основании анализа DFR-профилей 235 штаммов Y. pestis[1], одного штамма - Y. pseudotuberculosis и с учетом данных о ранее описанных 32 геномоварах[2] [Li et al., 2008] нам удалось подразделить все исследованные к настоящему моменту изоляты на 92 DFR-типа (Din = 0,687) (рис. 1). Согласно полученным результатам, на территориях стран СНГ и в Монголии циркулируют как минимум 52 геномовара. Большинство геномоваров вошли в состав трех кластеров: A, B и C (по Y. Li et al. [2008]), соответствующих основным ветвям, 1, 2 и 0, дендрограмм, построенных M. Achtman et al. [2004] на основании данных SNP- и IS100-типирования (рис. 2). В нашем исследовании вне кластеров остался африканский "полевочий" штамм subsp. angola – Angola, что может свидетельствовать как о его относительно древнем происхождении, так и об отсутствии среди анализируемых изолятов других филогенетически близких ему штаммов. Как и в публикации наших китайских коллег [Li et al., 2008], все штаммы биовара orientalis (24 штамма в составе 10 геномоваров) вошли в состав кластера A/1, все штаммы биовара medievalis (67 штаммов в составе 28 геномоваров) – в кластер B/2, а представители биовара microtus (неосновные подвиды Y. pestis [Li et al., 2009]) (84 штамма в составе 26 геномоваров), за исключением геномовара 85, представленного штаммом Angola, - в кластер C/0. Штаммы биовара antiqua (58 штаммов в составе 27 геномоваров) распределились по различным ветвям кластеров A/1, B/2 и C/0.
В состав кластера B/2 входят две основные ветви (рис. 1). Одна из ветвей соответствует ветви 2.ANT на рис. 2 и представлена в основном геномоварами: 10-13, 17, 26-28, 58, 62, циркулирующими на территориях Китая и Монголии, геномоваром 19 - основным DFR-типом в граничащем с Китаем и Монголией природном очаге 38 и минорным для очага 1, Монголии, Китая и Непала, а также минорными геномоварами 57 и 59 природного очага 1.
Вторая ветвь соответствует ветви 2.MED на рис. 2 и образована, в основном, DFR-типами, характерными для природных очагов стран СНГ. Она включает геномовар 15 - основной DFR-тип штаммов биовара medievalis, циркулирующих в природных очагах Средней Азии.

Рисунок 1. NJ-дендрограмма и DFR-профили 92 геномоваров Y. pestis
"gv" – геномовар; "bv" – биовар; "ANT" – antiqua (заглавные буквы обозначают, что геномовар описан ранее); "INT" - intermedium[3] ; "med" - medievalis; "mic" - microtus; "ori" - orientalis; "ssp" – подвид (обозначены первой буквой названий, а в случае subsp. angola3 – двумя первыми буквами); "no" – количество штаммов, проанализированных в нашей работе; "?" – нет данных.
Рисунок 2. Дендрограмма микроэволюции 21 штамма Y. pestis по данным SNP типирования [Derbise et al., 2010]
Все изученные нами штаммы биовара orientalis вошли в состав кластера A/1 и были выделены за пределами СНГ, Монголии и Китая. В этот же кластер вошли штаммы биовара antiqua, циркулирующие на территории Монголии (геномовар 01b (минорный DFR-тип для китайских очагов B1, C, D, G и K2) – четыре штамма, геномовар 62 – один штамм) и стран СНГ в природных очагах: 31 (основной для соседнего китайского очага A [Li et al., 2008] геномовар 04 – один штамм), 33 (геномовар 60, отличающийся от геномовара 04 отсутствием DFR18, – один штамм) и 37 (геномовар 61 – девять штаммов, геномовар 01b – один штамм). В состав этого кластера входят и штаммы геномовара 03 биовара intermedium, циркулирующие в китайском очаге B3. Кластер включает две основные ветви, одна из которых (DFR-типы: 07-09, 32-35, 56, 86-89) соответствует ветви 1.ORI на рис. 2, а остальные геномовары в составе второй ветви - 1.ANT.
Кластер C/0 формировали две основные ветви (рис. 1), одна из которых включала геномовар 90 (ветвь 0.PE2 на рис. 2), а вторая DFR-типы 14 и 80 (ветвь 0.PE4 на рис. 2).
В 0.PE2 ветвь, кроме DFR-типа 90 входили геномовары: 24 (минорный геномовар штаммов bv. antiqua subsp. pestis из китайского очага C; основной - для subsp. ulegeica (семь штаммов из Монголии) и минорный для subsp. caucasica (по одному штамму из очагов 5 и 7)); 63, 67, 70, 71, 74, 72, 76 (по одному штамму subsp. caucasica из очагов 6, 7, 4, 4, 39, 39, 5, соответственно); 64 (по одному штамму subsp. caucasica из очагов 6 и 7) и основной для очага 39 геномовар 73 (семь штаммов subsp. caucasica).
Ветвь 0.PE4 более разнообразна по входящим в нее подвидам. Сюда вошли геномовары 62, 23, 30 основного подвида биовара antiqua из Монголии и Китая. Большинство входящих в группу 0.PE4 штаммов биовара microtus обладало DFR-типом 14 - основным геномоваром китайских подвидов qinghaiensis и xilingolensis, который также оказался характерным для трех монгольских штаммов, относимых к подвиду altaica. В случае выявления близкого родства этих штаммов и с помощью других методов молекулярного типирования следует поставить вопрос о тщательной проверке всех коллекционных штаммов подвида altaica, выделенных в Монголии, на предмет их перевода в соответствующий внутривидовой таксон. Геномовар 80 – основной для штаммов подвида altaica, выделенных в очаге 36 (12 штаммов) и в Монголии (один штамм). К этому же геномовару относится и единственный в нашем исследовании представитель bv. talassica A-1820. Штамм Pestoides A относится к этому же DFR-типу и, вероятно, является представителем подвида altaica или talassica. Штаммы subsp. caucasica геномовара 69 циркулируют в очагах 4 (два штамма), 5 (два штамма) и 7 (один штамм). Остальные входящие в ветвь 0.PE4 геномовары представлены одним - двумя штаммами подвидов hissarica, altaica, caucasica и ulegeica.
Маловероятно, что совпадение DFR-типов (03 и 24) у штаммов из географически изолированных природных очагов и разных внутривидовых групп (рис. 1) связано с их заносом с одной территории на другую. Скорее это совпадение можно объяснить независимой утратой амплифицируемых локусов или участков "посадки" праймеров и даже ошибками, допущенными при проведении коллекционной работы со штаммами. Верификация полученных нами результатов с помощью других методов мультилокусного генотипирования (MLVA, CRISPR-типирование и др.) поможет ответить на эти вопросы.
На основании анализа MLVA-профилей 601 штамма Y. pestis[4] и одного штамма - Y. pseudotuberculosis нам удалось подразделить все исследованные к настоящему моменту изоляты на 430 MLVA25-типов (Din = 0,993), причем 352 из них описаны впервые с нашим участием или непосредственно нами. На территориях стран СНГ и в Монголии циркулируют как минимум 119 MLVA25-типов (проанализировано 139 и 23 штамма, соответственно).
На начальном этапе нашей работы был создан совместный с китайскими и французскими коллегами электронный каталог [Li et al., 2009] предложенных C. Pourcel et al. [2004] профилей 25 VNTR-маркеров Y. pestis, включающий данные о генотипировании 383 штаммов из природных очагов Китая, 38 изолятов из СНГ и Монголии [Li et al., 2009], а также ранее опубликованные данные о 180 штаммах из французской коллекции штаммов чумного микроба [Pourcel et al., 2004]. Глобальная кластеризация Y. pestis, полученная нами с помощью MLVA (рис. 3), соответствует данным предыдущих исследований.
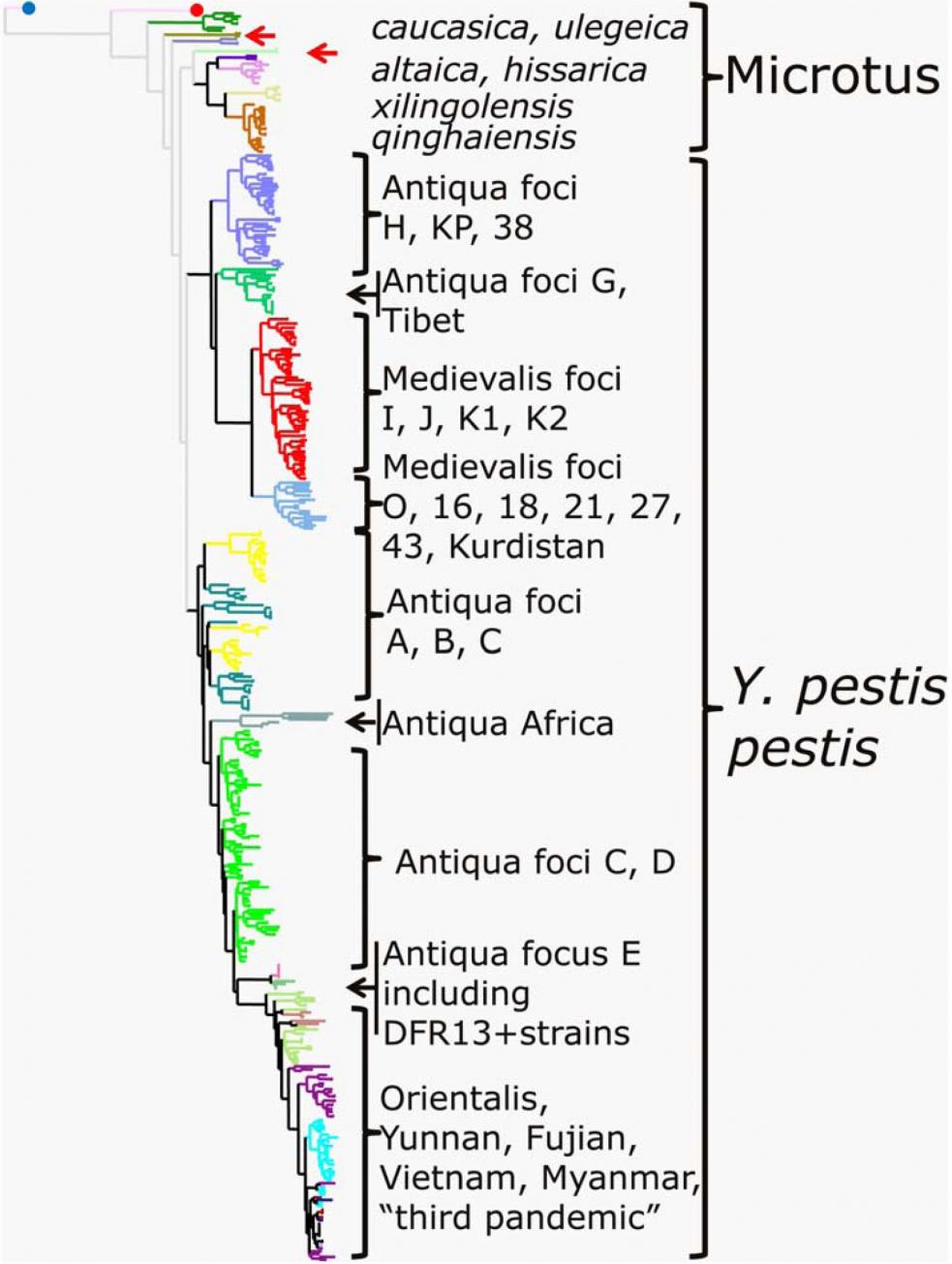
Рисунок 3. NJ-дендрограмма MLVA25-типов Y. pestis
Левая точка – "корневой вид" Y. pseudotuberculosis; правая точка - штамм Angola; стрелки - штаммы Y. pestis subsp. pestis, с неожиданными генотипами.
Так, SNP анализ выявил две отличающиеся по данным молекулярного типирования группы bv. antiqua, представляющие две линии Y. pestis (1.ANT и 2.ANT), эволюционно связанные с ветвями bv. orientalis и bv. medievalis [Achtman et al., 2004]. В этих ранних исследованиях линия 1.ANT была представлена африканскими изолятами bv. antiqua. Аналогичная кластеризация bv. antiqua была проведена с помощью CRISPR-типирования [Pourcel et al., 2005]. Эти две ветви были классифицированы как азиатская и африканская, но настоящее исследование показывает, что оба кластера bv. antiqua циркулируют в азиатских природных очагах чумы.
Работа, начатая совместно с лабораториями G. Vergnaud и R. Yang [Li et al., 2009], получила продолжение в рамках федеральной целевой программы "Национальная система химической и биологической безопасности Российской Федерации (2009-2013 годы)". Созданный в рамках международного сотрудничества электронный каталог [Li et al., 2009], включающий информацию о MLVA25-генотипах более 450 штаммов Y. pestis, а также методические материалы по проведению молекулярного типирования этим методом в 2009 г. были переданы нами для практического использования в Иркутский научно-исследовательский противочумный институт Сибири и Дальнего Востока и Ставропольский научно-исследовательский противочумный институт. Совместно был определен перечень штаммов для дальнейшего исследования, и часть из них была передана в ГНЦ ПМБ для проведения генотипирования в нашей лаборатории. Значительное увеличение количества анализируемых штаммов из регионов Кавказа и Горного Алтая дало более полную картину о биоразнообразии чумного микроба, циркулирующего в природных очагах, расположенных на этих территориях. Так, увеличение числа изолятов из очагов 4-7 с семи до 24 и включение в исследование девяти штаммов из очага 39 позволило выявить в составе кластера subsp. caucasica три независимые ветви (рис. 4.А), соответствующие природным очагам 39 (Дагестан), 4 (Армения и Грузия) и группе очагов 5-7 (Азербайджан и Армения). Полученные данные свидетельствуют, что очаги 5, 6 и прилегающую к ним часть очага 7 можно рассматривать как единый природный очаг чумы.
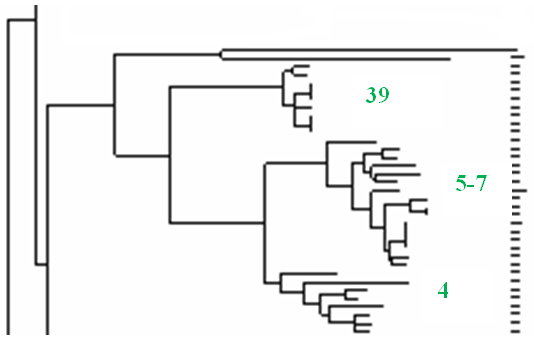 |  |
| А | Б |
Рисунок 4. Фрагменты NJ-дендрограммы MLVA25-типов штаммов Y. pestis (А) subsp. caucasica (цифрами указаны номера природных очагов чумы) и (Б) subspp. talassica, qinghaiensis, xilingolensis, hissarica и altaica
Увеличение количества штаммов subsp. ulegeica с трех до десяти не изменило положения этой относительно автономной гомогенной группы относительно других ветвей дендрограммы.
Штаммы bv. microtus из очагов 34, 36, 40, L, M и изоляты subsp. altaica из Монголии образовывали единый кластер, в одну из ветвей которого входили подвиды talassica, qinghaiensis, xilingolensis, а во вторую - hissarica и altaica (рис. 4.Б). Результаты MLVA подтверждают представленные в предыдущей главе данные о принадлежности штаммов I-3088, I-3132 и I-3134 не к подвиду altaica, а к xilingolensis.
При анализе штаммов биоваров antiqua и medievalis из природных очагов СНГ и Монголии также подтверждена приуроченность определенных MLVA25-типов к конкретным природным очагам чумы.
Построение достоверных деревьев филогенеза отдельных видов требует сочетанного использования информации о многих локусах/генах, полученной с помощью различных технологий (MLVA, SNP-, IS-, DFR-типирование и т.д.), и подбора аналитических методов, дающих близкие результаты кластеризации для различных технологий генотипирования [Heled, Drummond, 2010]. В результате использования опции "composite data sets" программы Bionumerics 5.10 мы получили очень “простую” и логичную дендрограмму-филограмму (рис. 5), в которой, в отличие от DFR-, MLVA- и SNP-дендрограмм нет проникновения генетических групп одного биовара в ветви, сформированные представителями других биоваров. По данным сочетанного DFR/MLVA25-типирования (Din = 0,996) наиболее древними представителями вида является группа изолятов биовара antiqua из Центральной Африки, за ними следует уникальный штамм 797 того же биовара, но выделенный по данным паспорта в среднеазиатском очаге 25, в котором циркулируют штаммы bv. medievalis. Скорее всего, этот штамм был занесен на территорию Туркменистана с приграничных территорий Ирана или Афганистана. Наиболее филогенетически близок к штамму 797 штамм Angola (bv. microtus subsp. angola), а затем следуют представители subsp. caucasica, азиатская ветвь биовара antiqua и т.д. (рис. 5). Вероятно, что проведение сочетанного анализа результатов генотипирования в формате DFR/MLVA/CRISPR/MLST/SNP позволит построить филограмму, приближающуюся по достоверности к результатам анализа данных полногеномного секвенирования.
Что же касается практического использования исследованных методов молекулярного типирования и созданного в ходе этого исследования электронного каталога DFR- и MLVA25-генотипов, то его можно продемонстрировать на примере внутривидовой идентификации штаммов Pestoides A и Pestoides F. После передачи группы штаммов из СССР в США их паспорта были утрачены, а затем присвоены новые наименования. Результаты MLVA25-типирования (DFR/MLVA25-типирования) однозначно свидетельствуют о том, что штамм Pestoides A был выделен в Горно-алтайском очаге 36 или на граничащей с ним территории Монголии, а штамм Pestoides F – в группе очагов Закавказского нагорья - 04-07.
[1] DFR-профили 165 штаммов определены в настоящем исследовании экспериментально, аналогичные данные о 48 штаммах взяты из работы [Li et al., 2008], а DFR-профили 22 штаммов получены в результате анализа полногеномных нуклеотидных последовательностей, доступных в Интернете.
[2] В работе [Li et al., 2008] первый геномовар включает два "подгеномовара": 1a и 1b.
[3] Аргументация по необходимости введения во внутривидовую классификацию новых таксонов приведена далее.
[4] MLVA25-профили 176 штаммов определены в нашей лаборатории, данные о 22 штаммах получены в результате анализа полногеномных нуклеотидных последовательностей, доступных в Интернете, а остальная информация взята из нашей совместной работы с зарубежными коллегами [Li et al., 2008].