
Методы масс-спектрометрии для решения стереохимических задач в ряду производных цитизина и циклопропанкарбоновой кислоты
На правах рукописи
ЕРАСТОВ АЛЕКСЕЙ СЕРГЕЕВИЧ
МЕТОДЫ МАСС-СПЕКТРОМЕТРИИ ДЛЯ РЕШЕНИЯ СТЕРЕОХИМИЧЕСКИХ ЗАДАЧ В РЯДУ ПРОИЗВОДНЫХ ЦИТИЗИНА И ЦИКЛОПРОПАНКАРБОНОВОЙ КИСЛОТЫ
02.00.04 - Физическая химия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата химических наук
Уфа – 2013
| Работа выполнена в Федеральном государственном бюджетном учреждение науки Институте органической химии Уфимского научного центра Российской академии наук. | ||
| Научный руководитель: Официальные оппоненты: | кандидат химических наук Галкин Евгений Григорьевич Кантор Евгений Абрамович доктор химических наук, профессор (ФГБОУ ВПО «Уфимский государственный нефтяной технический университет», заведующий кафедрой физики) Грабовский Станислав Анатольевич кандидат химических наук, доцент (ФГБУН Институт органической химии Уфимского научного центра РАН, старший научный сотрудник) | |
| Ведущая организация: | Федеральное государственное бюджетное учреждение науки Институт органической и физической химии им. А.Е. Арбузова РАН | |
Защита состоится 26 апреля 2013 г. в 14 00 часов на заседании диссертационного совета Д 002.004.01 в Федеральном государственном бюджетном учреждение науки Институте органической химии Уфимского научного центра Российской академии наук по адресу: 450054, Башкортостан, г. Уфа, проспект Октября, 71, зал заседаний. Телефакс: (347) 2356066. Е-mail: [email protected].
С диссертацией можно ознакомиться в научной библиотеке Уфимского научного центра РАН.
Автореферат разослан 23 марта 2013 г.
Ученый секретарь диссертационного совета
доктор химических наук, профессор Ф. А. Валеев
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность темы.
В последние годы современные методы масс-спектрометрии, такие как MALDI и электроспрей, находят всё большее число стереохимических приложений. Однако для многих объектов исследования наилучшим образом подходит классический метод хроматомасс-спектрометрии электронного удара, поскольку он позволяет анализировать смеси без предварительного разделения, а спектры являются наиболее информативными. Таким образом, изучение возможностей метода электронного удара в стререохимии представляется актуальным. В качестве объектов исследования выбраны два класса соединений – природные и синтетические хинолизидиновые алкалоиды, а также производные метилового эфира циклопропанкарбоновой кислоты. Производные хинолизидиновых алкалоидов привлекают внимание исследователей благодаря высокой нейрофизиологической активности. Внимание ко второй группе соединений вызвано проводимыми в ИОХ УНЦ РАН исследованиям реакций каталитического взаимодействия ненасыщенных соединений с метилдиазоацетатом. В обоих случаях возникают проблемы идентификации и оценки вкладов стереоизомеров в полученных реакционных смесях.
Работа выполнена в соответствии с планами научно-исследовательских работ Федерального государственного бюджетного учреждения науки Института органической химии Уфимского научного центра Российской академии наук по темам «Разработка новых методов синтеза гетероциклических систем» (номер государственной регистрации 0120.0 80144) и «Выделение, структурные исследования, трансформации, синтез и биологическая активность природных соединений (номер государственной регистрации 01201152194)
Цель работы.
Поиск масс-спектрометрического способа, позволяющего определить содержание диастереомеров хинолизидиновых алкалоидов в смеси независимо от содержания в ней посторонних компонентов; идентификация продуктов реакции взаимодействия метилдиазоацетата с ненасыщенными соединениями и определение их конфигурации.
Научная новизна и практическая значимость.
Впервые для определения содержания диастереомеров в их смеси применены масс-спектрометрические методики на основе сканирования электрического поля и сканирования выбранных ионов, что привело к увеличению различий между спектрами в диастереомерных парах и повышению чувствительности метода к целевым соединениям. Использование сканирования электрического поля в сочетании с ионной масс-фрагментографией позволило добиться улучшения разделения хроматографических пиков диастереомеров.
Применив методы хроматомасс-спектрометрии электронного удара и тандемную масс-спектрометрию, удалось найти способ идентификации продуктов взаимодействия ненасыщенных соединений с метилдиазоацетатом. С целью увеличения различия между практически одинаковыми масс-спектрами цис- и транс- изомеров полученных циклопропановых производных, применена техника ионизации при низкой энергии электронов, а проведённые ab initio расчёты позволили отнести масс-спектр к форме того или иного изомера. Такой комплекс экспериментальных и расчётных методов для решения подобного рода задач ранее не применялся.
Апробация работы.
Результаты работы представлены на Всероссийской конференции по органической химии (г. Москва, 2009 г.), Всероссийской научной INTERNET-конференции «Интеграция науки и высшего образования в области био- и органической химии и биотехнологии» (г. Уфа, 2011 г.), V Всероссийской конференции «Новые достижения в химии и химической технологии растительного сырья» (Барнаул, 2012 г.)
Публикации.
По материалам диссертации опубликованы 2 статьи в рецензируемых журналах, и тезисы 5 докладов.
Структура и объем диссертации.
Диссертационная работа изложена на 129 страницах, содержит 4 таблицы, 7 схем и 6 рисунков; состоит из введения, литературного обзора, обсуждения результатов, экспериментальной части, выводов, списка литературы и приложения. Список цитируемой литературы включает 144 наименований. В приложении приведены масс-спектры изучаемых соединений.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
- Определение диастереомерного состава производных N-(2-оксиэтил)-цитизина
Масс-спектрометрия электронного удара является удобным методом для анализа природных и синтетических алкалоидов хинолизидинового ряда. Для оценки вкладов диастереомеров в смесях производных N-(2-оксиэтил)-цитизина (1-6) необходимо было разработать масс-спектрометрический способ, работающий независимо от содержания посторонних компонентов, а также оценить его точность и границы применимости. Сложность заключалась в том, что масс-спектры диастереомеров на качественном уровне близки, а количественных различий не достаточно для анализа смеси обычным методом. Эффективного хроматографического разделения добиться не удавалось, поэтому вычислить вклад по площадям пиков не представлялось возможным.
Изучены 3 пары диастереомеров: N-(2-адамантил-2-оксиэтил)-цитизин 1 и 2, N-(2-фенил-2-оксиэтил)-цитизин 3 и 4, N-(2-оксипропил)-цитизин, 5 и 6. Для исследования были доступны как индивидуальные диастереомеры, так и реакционные смеси, содержащие R- и S- формы в разных соотношениях, а также посторонние примеси.
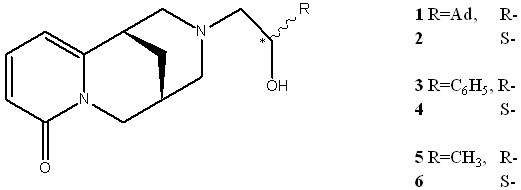
Масс-спектры соединений 1-6 характеризуются крайне малой интенсивностью молекулярных ионов. Поэтому определение брутто-состава с использованием масс-спектров высокого разрешения проводилось по значениям пиков ионов [M-H2O]+. Наиболее интенсивные пики в масс-спектрах соответствуют: отрыву заместителя R (m/z 233), отщеплению групп CH(OH)R или O=CHR (m/z 203 и 204 соответственно), а также ионам, образующихся при расщеплении гетероцикла по узловым точкам.
Масс-спектры всех шести компонентов на качественном уровне очень близки. Наиболее заметное различие в каждой паре диастереомеров – в относительной интенсивности ионов m/z 203 и 204. Разница для пары изомеров составляет 10-40%. В принципе такого различия в масс-спектрах должно быть достаточно для уверенного отнесения масс-спектра к какой-либо конкретной форме. Но при переходе к смеси изомеров появляется проблема их разделения. При хроматографировании оба диастереомера (для каждой из трёх пар) выходят широкими пиками с практически одинаковыми временами выхода. Масс-спектры накладываются друг на друга и регистрируются как один усреднённый. Использовать другие колонки не представляется возможным из-за слишком высокой температуры кипения соединений. Иными словами, при обычном хроматомасс-спектрометрическом анализе установить вклад изомеров в смеси не представляется возможным.
Для решения данной проблемы нами было предложено производить сканирование не полного масс-спектра, а только узкого диапазона масс, в который попадают интересующие ионы m/z 203 и 204. Вместо традиционного сканирования магнитного поля было решено использовать сканирование электрического поля. Данный способ позволяет более чем на порядок повысить чувствительность по интересующей нас паре ионов, а также понизить время записи спектра. Влияние посторонних компонентов практически исключено, поскольку даже если они образуют малоинтенсивные ионы, попадающие в выбранный диапазон, их наличие не помешает интерпретации, к тому же слишком мала вероятность того, что они попадут в этот же хроматографический пик.
Таким образом, поменяв режим регистрации спектра, хроматограмма существенно измененилась. Во-первых, практически полностью исчез уровень фона, во-вторых, пропали пики посторонних компонентов. Ну, а самый важный результат – на единственном оставшемся уширенном пике стало наблюдаться некоторое разделение. Программе обработке данных не составило труда вычислить вклад каждого диастереомера для каждой из трёх пар соединений.
Для оценки границ применимости предложенного способа анализа был проделан следующий опыт. Приготовлены три раствора с содержанием соединений 5 и 6 в соотношении 1:10, 1:1 и 10:1. Объём раствора подбирался таким образом, чтобы масса вещества составляла порядка 10-8 г. Во всех трёх случаях программе обработки данных удалось разделить пары полученных хроматографических пиков и вычислить вклад каждого из них. Для смеси 1:1 полученный результат очень близок к теоретическому. При малых концентрациях наблюдается отклонение в сторону уменьшения количества минорного компонента. При концентрации компонента 5 10% погрешность составляет 3%. Таким образом, метод не претендует на высокую точность, но в большинстве случаев точное значение не так важно. Главное его преимущество – возможность работать с низкими концентрациями, а также в присутствии большого числа посторонних соединений.
- Определение диастереомерного состава аддуктов реакции Дильса-Альдера, образованных при взаимодействии N-метилцитизина, (-)-леонтидина и (-)-термопсина с N-фенилмалеимидом
Каждый из трёх аддуктов существует в форме пары диастереомеров 7-8, 9-10 и 11-12. В нашем распоряжении были как чистые соединения, так и реакционные смеси.
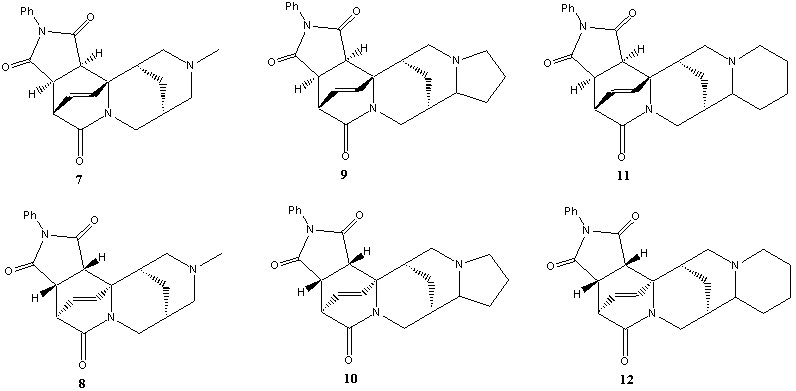
Масс-спектры данных соединений получены впервые. Они характеризуются довольно интенсивными пиками молекулярных ионов, а также двумя пиками ионов, образованными по ретрореакции Дильса-Альдера. Диастереомеры заметно отличаются между собой: вклад молекулярных ионов в полный ионный ток для соединений 7, 9, 11 значительно больше, чем у соответствующих изомеров 8, 10, 12.
Вследствие высокой температуры кипения использование газохроматографического разделения не представлялось возможным. Если при хроматографическом анализе удалось добиться хотя бы небольшого разделения по времени, то в случае прямого ввода надеяться на это не стоило. При использовании прямого ввода единственным критерием вклада содержания компонентов может стать отношение интенсивностей характеристических ионов. Чем точнее удастся его измерить, тем точнее можно провести количественный анализ.
В качестве характеристических ионов можно выбрать любую пару. Главное, чтобы в диастереомерах различие по ним было максимальным. Второе важное условие – необходимо, чтобы они находились вблизи друг друга, поскольку при сканировании электрического поля в большом диапазоне масс, чувствительность к более тяжёлым фрагментам заметно снижается. Выбор пал на ионы m/z 173 для всех соединений и ионов m/z 204, 230 и 244 для 7-8, 9-10 и 11-12 соответственно. Несмотря на то, что максимальное различие в спектрах проявляется в интенсивностях пиков молекулярных ионов, использовать их при данном способе анализа нецелесообразно, поскольку они находятся на большом удалении от других интенсивных ионов.
Для того чтобы как можно более точно измерить интенсивности интересующих ионов, сканирование проводилось в режиме регистрации выбранных ионов (SIM). Данный способ анализа обладает максимальной чувствительностью, доступной на магнитно-секторном приборе, так как сканируется не весь диапазон масс, а лишь несколько выбранных ионов. При каждом цикле сканирования прибор автоматически подстраивает параметры сканирования, сверяясь с парой реперных ионов стандарта (перфторкеросина). Благодаря высокому разрешению посторонние примеси практически не влияют на точность измерения. Данный метод прекрасно зарекомендовал себя в количественном анализе.
Искажения, вызванные изменяющимся ускоряющим напряжением, безусловно, присутствовали. Но поскольку эталонные спектры для индивидуальных диастереомеров регистрировались в тех же условиях, что и для смесей, ошибка в измерениях интенсивностей ионов особого значения не имела.
Вводимое в источник ионов количество вещества подбиралось таким образом, чтобы интенсивность регистрируемых ионов попадала в динамический диапазон масс-спектрометра. Экспериментально было выбрано значение порядка 10-8 – 10-6 г.
Для оценки случайной погрешности был выполнен следующий эксперимент. В режиме сканирования выбранных ионов зарегистрированы масс-спектры соединений 7 и 8 (в их спектрах различие по интенсивностям ионов максимальное). Количество второго на порядок превосходило количество первого (10-7 и 10-8 г). Из каждого опыта были выбраны по 30 сканирований, равномерно перекрывающих область выхода вещества. В расчёт не брались величины ионного тока, полученные на пороге чувствительности или «зашкалившие» значения. Также отбрасывались сканирования, явно выбивающиеся из общей зависимости. Величины отношения интенсивности иона m/z 204 к иону m/z 173 (%) представлены на графике (сх. 1).
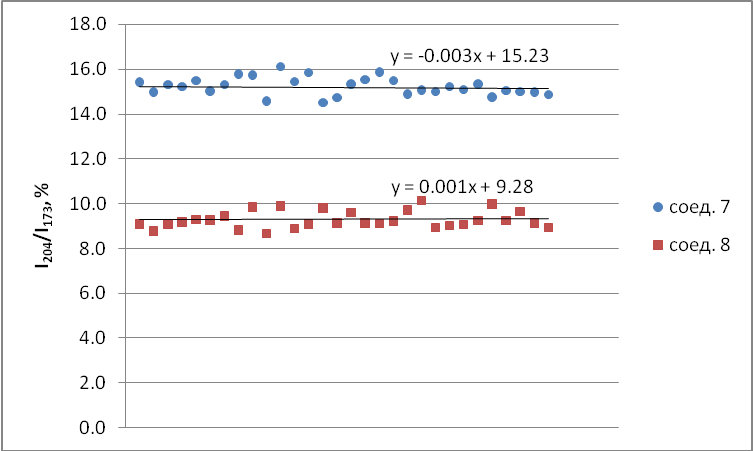
Схема 1. Усреднённые результаты отношения I204/I173 в 30-ти сканированиях для диастереомеров 7 и 8.
Видно, что точки хорошо ложатся на прямые, параллельные оси х. Рассчитанная по методу Стьюдента ошибка в измерении концентрации составила порядка 5%. Конечно, это число не учитывает другие факторы, которые могут повлиять на измерение, такие как фракционирование смеси (неодновременное испарение изомеров) и матричные эффекты (влияние посторонних компонентов).
Для определения нижнего предела чувствительности были приготовлены три смеси с концентрацией каждого диастереомера 10-3, 10-4 и 10-5 г/л. Для анализа отбирали по 1 мкл из каждого образца. То есть, в источник ионов вводили 210-9, 210-10 и 210-11 г смеси диастереомеров с соотношением 1:1. Рассчитанное значение относительной интенсивности I204/I173 во всех случаях должно составлять 12,25. Полученные значения вместе с доверительным интервалом представлены на графике (сх. 2).
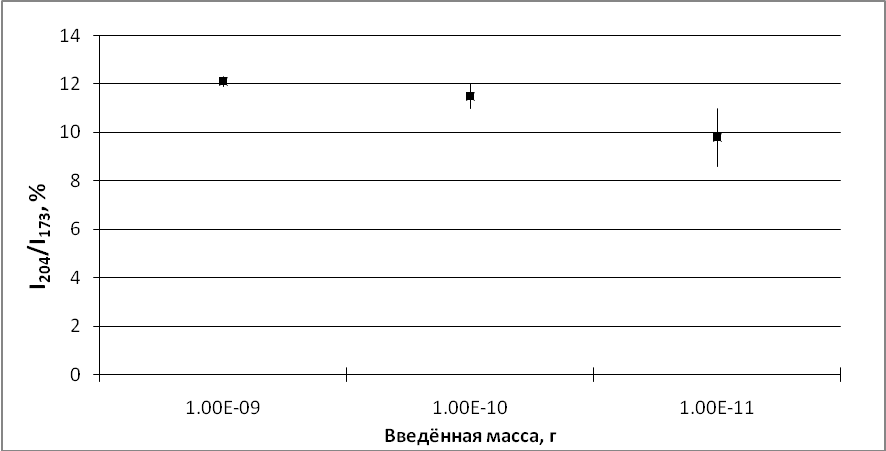
Схема 2. Точность анализа и доверительный интервал в зависимости от концентрации. Рассчитанное значение I204/I203 составляет 12.25.
Видно, что для первой смеси рассчитанное значение хорошо совпадает с экспериментальным, доверительный интервал вполне приемлемый. При дальнейшем снижении концентрации не только увеличивается разброс значений между различными сканированиями (что ухудшает доверительный интервал), но и проявляется ошибка, связанная с низким уровнем сигнала менее интенсивного иона m/z 204, приводящая к занижению измеренного значения. Предел обнаружения находится в области гораздо более низких концентраций, но при этом нельзя с уверенностью судить о вкладе стереоизомеров. Высокие концентрации также приводят к ошибке, связанной с «зашкаливанием» ионного тока максимального иона m/z 173, что приводит к завышению результата. Но эта проблема решается разбавлением раствора.
Таким образом, использование сканирования выбранных ионов с введением пробы через прямой ввод может найти практическое применение, когда необходимо определить состав смеси стереоизомеров, при условии, что они имеют хотя бы небольшое, но выраженное различие в масс-спектрах. Проведение анализа в режиме высокого разрешения зачастую лишает необходимости предварительной очистки вещества. Более того, возможно анализировать даже такие сложные смеси, как растительные экстракты, содержащие целевой компонент в минорных количествах. Использование же методов ВЭЖХ/МС также не обязательно даст положительный результат, поскольку метод ионизации электроспрей относится к мягким, и не вызывает глубокую фрагментацию; применение тандемных методов доступно отнюдь не на всех приборах.
Самое ценное, что метод не ограничивается анализом пары диастереомеров. Кроме них возможно анализировать другие структурные изомеры со схожими на качественном, но различными на количественном уровнях спектрами.
К недостаткам следует отнести необходимость правильно выбрать концентрацию, чтобы избежать выхода за рамки динамического диапазона детектора. Необходимо располагать хотя бы небольшим количеством очищенных изомеров, чтобы выбрать из их обычных масс-спектров нужные ионы и зарегистрировать спектры в режиме SIM. Необходимо также быть уверенным, что выбранные ионы не образуются другими соединениями, содержащимися в смеси. Ещё один минус – анализ целевой, с его помощью нельзя ничего узнать о других, в том числе и об основных компонентах смеси.
- Определение принадлежности к циклопропановой или открытой форме продуктов каталитического взаимодействия метилового эфира диазоуксусной кислоты с ненасыщенными соединениями
В лаборатории металлоорганического синтеза и катализа ИОХ УНЦ РАН проводилось изучение каталитического взаимодействия ненасыщенных соединений с метилдиазоацетатом. Нашей целью было масс-спектрометрическое исследование полученных реакционных смесей.
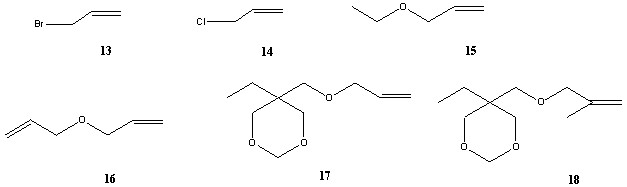
В каталитической реакции из соединений 13-18 образуются не только производные метилового эфира циклопропанкарбоновой кислоты (19-24), но и изомерные им продукты с двойной связью (25-30), а также более высокомолекулярные соединения, образованные при вторичной реакции (сх. 3).
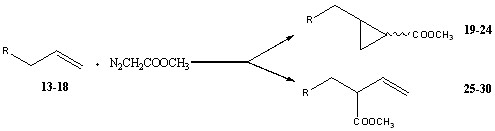 Схема 3. Два основных направления протекания реакции взаимодействия ненасыщенных соединений с метилдиазоацетатом.
Схема 3. Два основных направления протекания реакции взаимодействия ненасыщенных соединений с метилдиазоацетатом.
Количество продуктов и их выходы сильно различаются в зависимости от природы олефина, катализатора и условий реакции. Кроме того, каждое из соединений 19-24 может существовать в форме цис- и транс- изомеров (19-24)а и (19-24)б. Таким образом, на хроматограмме может наблюдаться немалое число пиков.
Выполнить их идентификацию – сложная задача, поскольку среди них наблюдается набор соединений с одинаковым составом и очень близкими масс-спектрами (что характерно для большинства изомеров, в частности циклопропанов). Вероятность ошибочной идентификации недопустимо высока. Использование других физико-химических методов, в частности ЯМР, связано со сложностью разделения смеси и наработкой нужного количества индивидуальных компонентов.
Масс-спектры, полученные при стандартном способе проведения анализа не содержат достаточной информации, позволяющей различить изомеры (19-24) и (25-30). Необходимо было найти способ, позволяющий с уверенностью определить наличие структурного фрагмента того или иного типа. Для этой цели наиболее удобно использовать метод тандемной масс-спектрометрии (диссоциация индуцированная столкновениями B/E=const). Он позволяет зарегистрировать масс-спектрометрическую характеристику не всего соединения, а любого выбранного нами иона. Полученный таким образом масс-спектр называют спектром дочерних ионов. Он показывает, какие ионы образовались из данного выбранного иона. Для иона каждой структуры характерен свой уникальный масс-спектр дочерних ионов. Различия в спектрах дочерних ионов будут свидетельствовать о различии в структурах родительских ионов. Таким образом, для того чтобы установить принадлежность соединения к какому-либо классу, необходимо получить масс-спектры дочерних ионов известных соединений, содержащих данный характеристический фрагмент.
Характеристический фрагмент должен отвечать следующим требованиям: присутствовать во всех соединениях, характеризоваться большой интенсивностью, иметь простой и понятный механизм образования, он должен подвергаться дальнейшему распаду (для того чтобы его спектр состоял не из единственного иона), а также быть общим для стереоизомеров. Всем этим требованиям удовлетворяет ион m/z 113, имеющий брутто-состав C6H9O2+ (подтверждено данными масс-спектрометрии высокого разрешения). В первом приближении его образование можно рассматривать как простую реакцию отщепления радикала (схема 4).
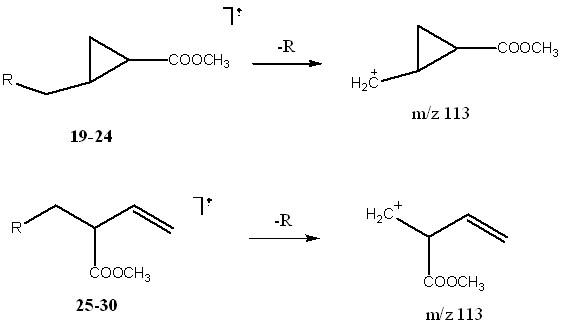
Схема 4. Условные структуры и механизм образования ионов 113 из циклической и открытой форм.
В действительности механизм реакций может быть сложнее, да и структуры ионов m/z 113 иные, чем на схеме, но в рассматриваем случае это не играет решающей роли. Важно то, что данные ионы характеризуется различными спектрами дочерних ионов, которые позволяет отличить циклическую форму от алифатической.
Для получения спектров CID структур с известным строением наилучшим образом подошла реакционная смесь, полученная из бромистого аллила (13). Она состоит из исходного соединения, двух пар геометрических изомеров 19 и 25 и небольшого количества примесей.
На хроматограмме, полученной в режиме связанных сканирований B/E=const для иона m/z 113, наблюдалось четыре пика. В каждой паре геометрических изомеров спектры с точностью повторялись, но несколько отличались между парами.
На рисунке 1 изображены полученные спектры B/E=const. На первый взгляд, различия могут показаться случайными. Тем не менее, они повторялись при повторном проведении опыта, и самое главное – в других реакционных смесях 14-17. Таким образом, необходимый признак, позволяющий различить циклопропановые структуры и их алифатические изомеры, был найден.
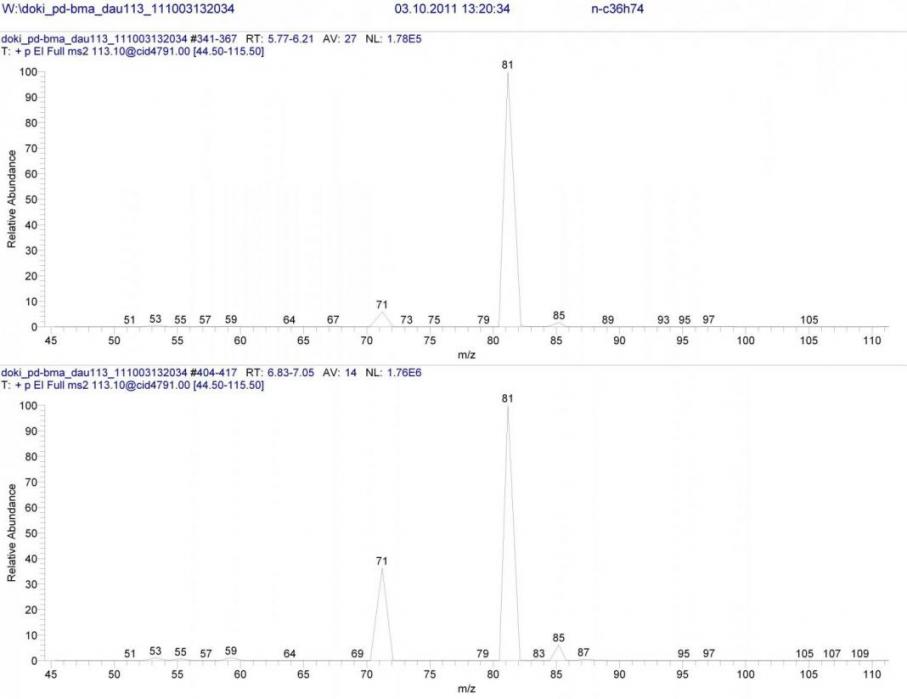 Рисунок 1. Масс-спектры B/E=const для ионов m/z 113 открытой и циклической форм.
Рисунок 1. Масс-спектры B/E=const для ионов m/z 113 открытой и циклической форм.
В реакционной смеси соединения 18 среди продуктов присутствуют только циклопропановые структуры. Поскольку в исходном ненасыщенном углеводороде атом углерода при двойной связи замещён метильной группой, вместо иона m/z 113 необходимо рассматривать гомологичный ион m/z 127. Безусловно, спектр его дочерних ионов будет другим, но общие черты фрагментации оказались весьма схожие (рис. 2).
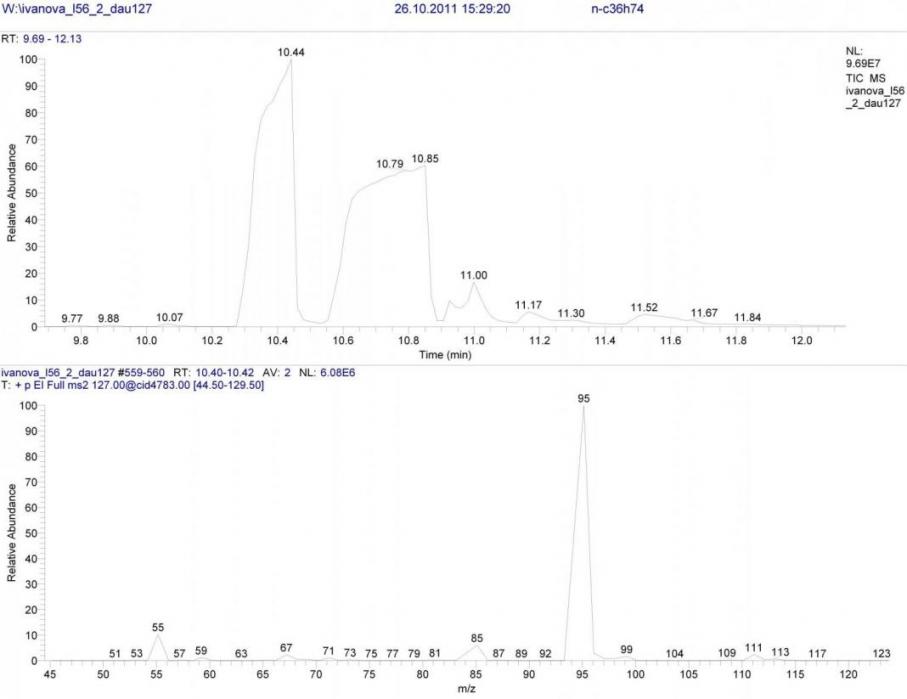 Рисунок 2. Масс-спектр B/E=const для иона m/z 127 циклической формы.
Рисунок 2. Масс-спектр B/E=const для иона m/z 127 циклической формы.
Основные ионы в данном спектре либо гомологичны (m/z 95, 99), либо такие же, как в спектре иона m/z 113 (m/z 85).
Данный подход может использоваться при анализе смесей, образованных в сходных реакциях. Для этого потребуется предварительно создать библиотеку спектров дочерних ионов возможных структурных фрагментов, что в дальнейшем значительно упростит процесс отнесения спектра к определённому соединению.
- Стереоизомерное отнесение производных метилового эфира циклопропанкарбоновой кислоты
Вторая, более сложная задача, которая ставилась перед нами – предложить метод определения пространственного расположения заместителей в циклических структурах, пригодный для анализа реакционных смесей.
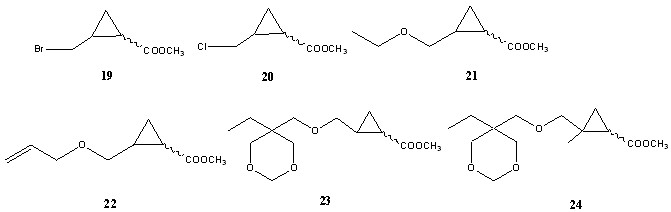
Соединения 19-24 могут существовать в форме цис- (а) и транс- (б) изомеров. Масс-спектры изомеров либо довольно близки друг к другу (19-22), либо различия имеют малопонятный, несистемный характер (23-24).
Как уже упоминалось ранее, основная причина таких отличий – высокая избыточная энергия молекулярных ионов, образовавшихся при ионизации электронами с энергией 70 эВ. Несмотря на то, что ион приобретает сверх потенциала ионизации относительно небольшое по сравнению с 70 эВ количество энергии, этого различия оказывается достаточно для протекания энергетически маловыгодных процессов, такие как перегруппировки в МИ и вторичная фрагментация. Совокупность этих процессов приводит к появлению большого количества ионов, интенсивности которых будут зависеть от многих факторов, и не могут быть однозначно интерпретированы. Само собой, в большинстве случаев, такие явления как стереоизомерные эффекты остаются скрытыми. Даже если они проявятся в виде различия интенсивностей каких-либо ионов, то далеко не всегда удаётся найти какие-либо закономерности, как это было в случае хинолизидиновых производных 7-12.
Для того чтобы избавиться от влияния побочных процессов энергия ионизирующих электронов была снижена со стандартной величины 70 эВ до величины, несколько превышающей потенциал ионизации исследованных соединений.
Влияние ионизирующего напряжения на масс-спектр было изучено на примере соединений 20(а, б). При величине 20 эВ наблюдалось только исчезновение малоинтенсивных ионов, очевидно, образованных в невыгодных процессах. При дальнейшем понижении энергии ионизации масс-спектр начинал меняться, снижалась интенсивность некоторых ионов. Одновременно с этим резко падала чувствительность прибора, поэтому, при величинах ниже 12 эВ, спектры уже не могли быть уверенно зарегистрированы даже при соответствующей подстройке параметров прибора. Относительная интенсивность молекулярного иона напротив возрастала. Из-за большого разброса энергий электронов нельзя добиться таких условий, чтобы в масс-спектре остался только молекулярный ион и несколько самых выгодных фрагментных. Однако, сравнивая масс-спектры при разных значениях энергии ионизации, можно выявить тенденцию к повышению или понижению относительной интенсивности ионов. Для соединения 20 с понижением энергии ионизации возрастает только относительная интенсивность МИ и иона m/z 113.
Наиболее удобным оказалось значение 12 эВ, так как при нём основные тенденции к изменению масс-спектров уже проявляются, но чувствительности прибора ещё вполне достаточно. Спектры всех остальных соединений 20-24 были зарегистрированы при данной величине. Как показали квантово-химические расчёты, потенциалы ионизации для всех соединений находились в пределах 6-8 эВ.
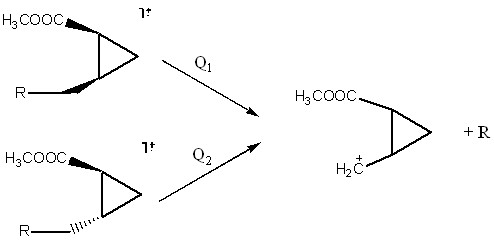
При 12 эВ масс-спектры стереоизомеров начинают заметно различаться по интенсивности пика иона m/z 113. Поскольку при низком значении ионизирующей энергии имеют место только наиболее выгодные процессы фрагментации, логично утверждать, что интенсивность иона m/z 113 будет больше в спектре того изомера, для которого процесс отщепления заместителей более выгодный. Поскольку реакции отщепления в случае обоих изомеров приводит к одним и тем же ионам и нейтральным фрагментам, наиболее выгодным этот процесс будет для изомера, обладающего наибольшей энтальпией образования (сх. 5).
![]()
Схема 5. Термодинамическое объяснение стереоизомерного эффекта в масс-спектрометрии
Это хорошо известный факт в масс-спектрометрии. Он нашёл своё подтверждение на многих соединениях даже при 70 эВ. Понизив напряжение ионизации, мы только лишь избавляемся от посторонних конкурирующих процессов, тем самым, делая вывод более надёжным.
Для того чтобы это правило работало, необходимо выполнения нескольких условий. Самое главное из них – оба изомера при распаде должны образовывать одинаковые продукты. Идентичность спектров B/E=const для иона m/z 113 даёт положительный ответ на этот вопрос. Второй важный момент. Масс-спектрометрический распад с точки зрения кинетики – сложная система параллельных и последовательных реакций. Мы же из этих реакций рассматриваем только одну. Само собой, конкурирующие процессы не могут не оказать на неё влияния. Несмотря на то, что пара стереоизомеров имеет общие пути фрагментации, большая часть которых не затрагивает центр, отвечающий за стереоизомерные различия, пренебрегать этими процессами не всегда корректно, а как-либо учесть не представляется возможным. Единственный выход – рассматривать только те соединения, в чьих спектрах ионы, образованные в посторонних процессах, мало интенсивны. Мы же пошли по другому пути – понизили интенсивность протекания посторонних процессов путём понижения энергии ионизирующих электронов.
В соответствии с вышеизложенным, отнесение хроматографического пика к цис- или транс- изомеру можно выполнить на основании одной лишь интенсивности иона m/z 113 в его низковольтном спектре. Тот спектр, в котором ион будет более интенсивен, должен соответствовать энергетически менее выгодной структуре. Для того чтобы определить, какая структура наиболее выгодна, мы применили квантово-химические расчёты.
Расчёты включали в себя поиск наиболее выгодного конформера для каждого из изомеров. Они проводились в три этапа с последовательным увеличением уровня расчётного метода. На каждом этапе проверялось принадлежность найденной конфигурации к локальному минимуму или седловой точке; отбрасывались заведомо невыгодные структуры. В целях экономии компьютерного времени на последнем этапе (самый точный метод) расчёт матрицы Гесса проводился только для самых выгодных структур; оставшиеся структуры не рассматривались. Для каждого изомера изначально подбиралось 4-6 гипотетических конформеров. Для соединений 23, 24 их количество возросло до 11. Чтобы избежать перебора большого количества конформеров, некоторые параметры, такие как, например, ориентация карбоксиметильной группы относительно циклопропанового кольца, определялись единожды и использовались в последующих расчётах.
Результаты расчёта величин разницы полных энергий транс-изомера и цис-изомера приведены в таблице 3.3. В первом случае расчёт проводился по методу MP2/6-311G(d,p), во втором по методу B3LYP/6-311G(d,p). Конформационный анализ при использовании любого метода выявлял одну и ту же наиболее выгодную структуру, но для большинства молекул конформационное состояние цис- и транс-изомеров оказывалось различным. Эта одна из причин больших различий по энергиям для некоторых структур, например, 21 и 22. Несмотря на то что на количественном уровне результаты расчётов с использованием разных методов сильно разнятся, ответ на главный вопрос остаётся одинаковым: во всех случаях цис- изомер менее выгоден, чем транс-изомер. Какой метод заслуживает большего доверия – вопрос открытый, поскольку сравнение с экспериментальными данными не проводилась. Наиболее правдоподобными выглядят результаты, полученные по методу B3LYP/6-311G(d,p).
| соед. | E(T)-E(С), кДж/моль | |
| MP2/6-311G(d,p) | B3LYP/6-311G(d,p) | |
| 19 | 0.5 | 3.1 |
| 20 | 0.5 | 2.8 |
| 21 | 3.4 | 6.1 |
| 22 | 11.8 | 10.9 |
| 23 | 2.2 | 5.5 |
| 24 | - | 2.1 |
Таблица 1. Результаты расчёта разницы полных энергий транс- и цис- изомеров методами MP2/6-311G(d,p) и B3LYP/6-311G(d,p).
Таблица 1 составлена по результатам расчётов только самых выгодных конформеров. Положительные значение величин разности энергий свидетельствуют о том, что транс-изомеры во всех случаях более выгодны. Ожидалось, что в случаях 23 и 24 из-за наличия внутримолекулярной водородной связи в цис-изомере и её отсутствии в транс-изомере картина может поменяться на противоположную, но этого не произошло. Несмотря на отсутствие значительных различий по энергии, структуры изомеров 23 и 24 различаются между собой гораздо больше, чем остальные пары изомеров. Такое различие в структурах не может не сказаться на предпочтительных направлениях фрагментации. При энергии электронов 70 эВ в спектрах наблюдается большое количество ионов, значительная часть которых обусловлена фрагментацией с участием диоксолановых циклов. Каждый из каналов фрагментации зависит не только от стереохимии, но и от протекания других конкурирующих процессов, которые, возможно, также зависят от стереохимии. То есть мы имеем очень сложную кинетическую систему. Однако при снижении энергии ионизирующего напряжения вклад конкурирующих процессов заметно убывает, но всё равно остаётся довольно высоким; это заметно снижает надёжность изомерного отнесения. И чем сложнее структура, тем менее достоверно её отнесение к определённому изомеру предложенным нами методом.
Для более простых соединений (19-22), напротив, количество конкурирующих процессов в низковольтной фрагментации заметно меньше, а разница по энергиям между цис- и транс- изомерами более значительная, так что их идентификация не вызывает больших сомнений.
Итак, к менее энергетически выгодному геометрическому изомеру следует отнести соединение, в масс-спектре которого вклад фрагментного иона m/z 113 больше, чем в масс-спектре его антипода. Естественно, берётся в расчёт интенсивность пика, выраженная по отношению к интенсивности какого-либо другого пика. В большинстве работ – к пику молекулярного иона. Однако наши соединения интенсивных молекулярных ионов не образуют. В низковольтных спектрах, они практически не регистрируются, вместо них наблюдаются ионы [M-H]+ и [M+H]+. Поэтому учитывая большое сходство спектров изомеров, величина характеристического иона m/z 113 была отнесена к полному ионному току (сумма интенсивностей всех ионов в спектре). Результаты измерений представлены в таблице 2. Полученная характеристика и послужила отправной точкой для стереоизомерных отнесений.
| соед. | Вклад иона m/z 113 в ПИТ, % | |
| Цис-изомер | Транс-изомер | |
| 19 | 41 | 30 |
| 20 | 52 | 34 |
| 21 | 14,8 | 11,7 |
| 22 | 28 | 15 |
| 23 | 13,1 | 10,7 |
| 24 | 20,3 | 17,5 |
Таблица 2. Вклады в полный ионный ток иона m/z 113 для цис- и транс- изомеров.
Из таблицы видно, что, как и ожидалось, отнесённая к полному ионному току интенсивность иона m/z 113 для цис- изомера всегда выше, чем для транс-изомера. Несмотря на полученное сходство экспериментальных данных с теоретическими предположениями, было решено более детально изучить механизм реакции элиминирования. Дело в том, что этот процесс может оказаться сложнее, чем одноактный простой разрыв связи. В таком случае, вывод о скорости протекания реакции, сделанный на основе термодинамических характеристик молекулярного иона и его фрагментов окажется некорректным.
- Изучение механизма распада производных метилового эфира циклопропанкарбоновой кислоты
Квантово-химические расчёты проведены для наиболее простых соединений 20(а, б). Если для 20б (транс-изомер) структура молекулярного иона принципиально не отличалась от структуры исходной молекулы, то для 20а (цис-изомер) картина была уже иная. При ионизации последнего наблюдается выгодный процесс переноса атома водорода от метиленовой группы к карбонильной с образованием дистонического иона. Для транс-изомера данный процесс невозможен. Однако при элиминировании радикала хлора снова наблюдается обратный перенос протона (вывод подтверждён расчётным методом релаксационного сканирования). В итоге, структура фрагментного иона m/z 113 остаётся такой же для цис- и транс- формы, но механизмы его образования существенно различаются (сх. 6).
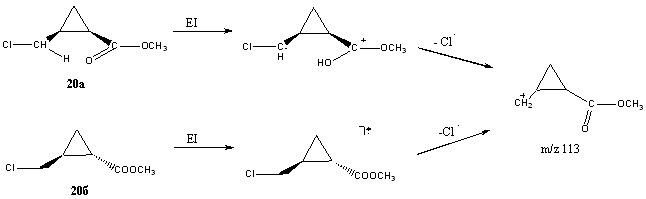
Схема 6. Различные механизмы отщепления радикала хлора для цис- и транс- изомера соединения 20.
Энергетические характеристики частиц изображённых на схеме 6, представлены на рисунке 3.
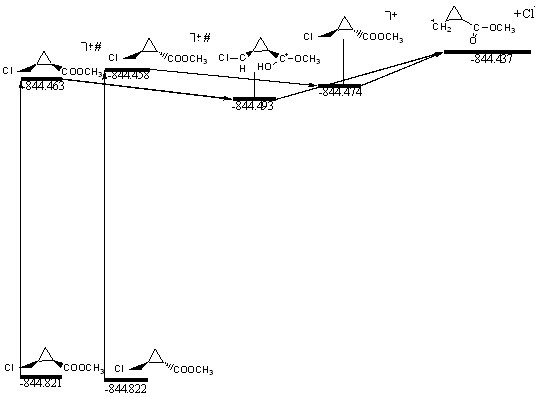
Рисунок 3. Энергетические характеристики процесса отщепления радикала хлора от молекул 20(а, б). Величины полной энергии указаны в Хартри. Знак # обозначает нерелаксированный молекулярный ион.
Первый акт ионизации - образование нерелаксированного молекулярного иона – частицы с геометрическими характеристиками, идентичными исходной молекуле, но лишённой одного электрона (на диаграмме отмечен знаком #). Такая частица является неустойчивой. Она стремится либо перейти в равновесное состояние, либо диссоциировать. Время перехода в равновесное состояние имеет порядок времени колебания связи 10-13 с. Это очень быстрый процесс, он не может заметно влиять на результирующую скорость распада. После перехода в равновесное состояние следует этап диссоциации. Поскольку, как было показано выше, в цис- изомере наблюдается водородный перенос, полученная структура оказывается более выгодной. Следовательно, её распад – менее выгодный процесс, чем для транс-формы. Из этого утверждения следует, что для цис- изомера процесс элиминирования радикала хлора менее выгодный; интенсивность иона m/z 113 должна быть ниже.
Полученное несоответствие с экспериментальными данными можно объяснить тем, что по каким-либо причинам данная схема не реализуется. Либо за счёт избыточной энергии диссоциация происходит до наступления релаксации, либо структура с переносом протона в реальности не образуется. Заметим, что для её образования требуется время, сравнимое с временем для диссоциации. Нельзя исключать также влияние конкурирующих процессов распада, которые данная схема никак не рассматривает. Тем не менее, из расчётов всё же можно заключить, что диссоциация стереоизомеров может протекать по совершенно разным механизмам.
ВЫВОДЫ
- Разработан способ определения содержания диастереомеров производных N-(2-оксиэтил)-цитизина в их смеси методом ГХ-МС в режиме сканирования электрического поля. Благодаря тому, что при данном методе сканирования за короткое время регистрируется лишь узкий диапазон масс, заметно возросла чувствительность и уменьшилось влияние посторонних примесей, а разделение хроматографических пиков соответствующих диастереомеров при том же хроматографическом режиме стало более выраженным.
- Для определения вкладов диастереомеров аддуктов реакции Дильса-Альдера, образованных по реакции N-метилцитизина, (-)-леонтидина и (-)-термопсина с N-фенилмалеимидом, использован метод сканирования выбранных ионов (SIM) с прямым введением пробы в источник ионов. Содержание диастереомеров определялось по величине отношения интенсивностей характеристических ионов, измеренной с большой точностью благодаря выбранному методу сканирования.
- При анализе продуктов взаимодействия ненасыщенных соединений с метилдиазоацетатом показана эффективность техники тандемной масс-спектрометрии. Поскольку полученные при этом спектры дочерних ионов для производных циклопропанкарбоновой кислоты и их изомеров с линейной цепью отличались друг от друга, идентификация соответствующих соединений заметно упростилась.
- С помощью техники низковольтной масс-спектрометрии, а также квантово-химических расчётов выполнено отнесение масс-спектров производных метилового эфира циклопропанкарбоновой кислоты к формам цис- или транс- изомеров. Если при традиционном проведении эксперимента масс-спектры изомеров не имели выраженных различий, то с применением низковольтной техники эти отличия приобрели закономерный характер, связанный с различиями во внутренних энергиях изомерных пар.
- Показано, что вследствие внутримолекулярных взаимодействий масс-спектрометрический распад диастереомерной пары метилового эфира 2-(хлорметил)циклопропанкарбоновой кислоты протекает по различным механизмам: с прямым разрывом связи для транс- изомера и с образованием перегруппировочного дистонического иона для цис- изомера.
Основное содержание работы изложено в следующих публикациях:
- Ерастов А.С., Галкин Е.Г. Структурный анализ изомерных компонентов смесей, образованных при реакции циклопропанирования метилдиазоацетатом. // Башкирский химический журнал. – 2012. – Т. 19. – № 1. – С.46-48.
- Ерастов А.С., Галкин Е.Г., Цыпышева И.П., Абдуллин М.Ф. Анализ вкладов диастереомеров производных цитизина методом масс-спектрометрии электронного удара с использованием прямого ввода и сканирования выбранных ионов. // Бутлеровские сообщения. – 2012. – Т.31. – №7. – С.38-42.
- Пташко Д.О., Ерастов А.С., Ханова М.Д., Галкин Е.Г. Особенности масс-спектрометрической фрагментации (циклопропилметил)фенилового эфира и (циклопропилметил)фенилового амина. // Тезисы докладов Всероссийской конференции по органической химии. – Москва. – 2009. – С.179.
- Пташко Д.О., Ерастов А.С., Ханова М.Д. Перегруппировка (циклопропилметил)-фенилового эфира в присутствии кислот Льюиса. // Тезисы докладов Всероссийской конференции по органической химии. – Москва. – 2009. – С.350.
- Пташко Д.О., Гайсин Р.Д., Ерастов А.С. Синтез и превращения циклопропансодержащих производных фенола и анилина. // Тезисы докладов Международной научно-технической конференции «Китайско-российское научно-техническое сотрудничество. Наука – Образование - Инновации». – Урумчи, КНР. – 2009. – С.76.
- Пташко Д.О., Ерастов А.С., Галкин Е.Г. Применение низковольтной масс-спектрометрии для установления конфигурации производных метиловых эфиров циклопропанкарбоновой кислоты. // Материалы VI Всероссийской научной INTERNET-конференции «Интеграция науки и высшего образования в области био- и органической химии и биотехнологии». – Уфа. – 2011. – С. 25-27.
- Ерастов А.С., Галкин Е.Г. Определение вкладов диастереомеров хинолизидиновых адкалоидов, модифицированных по реакции Дильса-Альдера методом масс-спектрометрии электронного удара. // Материалы V Всероссийской конференции с международным участием «Новые достижения в химии и химической технологии растительного сырья. – Барнаул. – 2012. – С.157.